Cell, Vol. 61,1349-1357, June 29, 1990, Copyright © 1990 by Cell Press
Jan C. Semenza, Kevin G. Hardwick, Neta Dean, and Hugh R.
B. Pelham
MRC Laboratory of Molecular Biology
Hills Road
Cambridge CB2 2QH
England
Summary
Resident proteins of the ER lumen carry a specific tetrapeptide signal (KDEL or HDEL) that prevents their secretion. We have previously described the isolation of yeast mutants that fail to retain such resident proteins within the cell. Here we describe ERD2, a gene required for retention. It encodes a 26 kd integral membrane protein whose abundance determines the efficiency and capacity of the retention system. Reduced expression of ERD2 leads to secretion of proteins bearing the HDEL signal, whereas overexpression of ERD2 improves retention both in wild-type cells and in other mutants. These results are consistent with other evidence that ERD2 encodes the HDEL receptor (see accompanying paper). The gene is also required, perhaps indirectly, for normal protein transport through the Golgi, and hence for growth. We discuss possible roles for ERD2 in the secretory pathway.
Introduction
In eukaryotic cells, newly synthesized secretory proteins are first inserted into the endoplasmic reticulum (ER) and are then transported in vesicles to the Golgi and eventually to the cell surface (Palade, 1975). However, such proteins comprise only a small fraction of the total protein content of the ER lumen. The most abundant polypeptides are resident soluble proteins such as BiP (a member of the hsp70 family), grp94 (a relative of hsp90), and protein disulfide isomerase (for review see Pelham, 1989). The main role of these proteins seems to be to aid the translocation, folding, and maturation of secretory proteins.
Transport of secretory proteins from the ER to the cell surface appears to occur in a nonselective fashion: no specific transport signal has yet been identified (Pfeffer and Rothman, 1987; Rose and Doms, 1988). In contrast, the soluble residents of the ER are distinguished by a C- terminal tetrapeptide sequence, which in animal cells is usually KDEL. This sequence is both necessary and sufficient for retention in the ER (Munro and Pelham, 1987; Pelham, 1988; Zagouras and Rose, 1989; Mazzarella et al., 1990).
Proteins bearing the KDEL signal are not immobilized in the ER. Examination of the carbohydrate modifications that occur to a KDEL-tagged lysosomal enzyme, cathepsin D, suggests that they can be transported to a subsequent compartment on the secretory pathway by the same nonselective transport vesicles that carry proteins destined for secretion (Pelham, 1988). ER proteins are then thought to be recovered by a receptor-mediated process. They are assumed to bind to a KDEL receptor in a "salvage compartment" (which in animal cells probably corresponds to the "15° C compartment") and to be incorporated into specialized vesicles that carry them back to the ER (Pelham, 1989).
To identify essential components of this recycling pathway, we have used a genetic approach. Our strategy has been to isolate mutants of the yeast Saccharomyces cerevisiae that are defective in the retention system. Previous work has established that yeast uses HDEL, rather than KDEL, as an ER retention signal; that HDEL- containing proteins can leave the ER, receive Golgi-specific carbohydrate modifications, and then return; and that the retrieval system is saturable, as expected for a receptor- mediated process (Pelham et al., 1988; Dean and Pelham, 1990). Extensive mutagenesis resulted in the identification of two genes required for the retention in the ER of HDEL- tagged invertase fusion proteins; these were named erd1 and erd2 (for "ER retention defective"). We have recently shown that deletion of the ERD1 gene causes a pleiotropic defect in part of the Golgi apparatus, which evidently results in inefficient retrieval from this organelle (Hardwick et al., 1990).
In this paper we describe the characterization of the ERD2 gene. This gene encodes an integral membrane protein that is required both for retention of ER proteins and, perhaps indirectly, for normal traffic of proteins through the Golgi. Strikingly, the capacity of the retention system is determined by the level of expression of the ERD2 gene. This observation, together with evidence that ERD2 determines the signal specificity of the retention system (Lewis et al., 1990 [accompanying paper]), leads us to propose that ERD2 encodes the HDEL receptor.
Results
erd2 Mutants Secrete BiP
The erd2 mutants were selected for their ability to secrete
an invertase fusion protein bearing the HDEL signal (Pelham et
al., 1988; Hardwick et al., 1990). To see whether an endogenous
ER protein was also secreted, an erd2 strain was pulse-labeled
with 35SO4 and cells and media were subjected
to immunoprecipitation with an antibody specific for BiP. As a
control, we also examined a strain (YFGR) in which the BiP gene
had been modified such that it encoded a protein with the C terminal
sequence FGR instead of FEHDEL (Hardwick et al., 1990). Since
it lacks the HDEL signal, this protein is not a substrate for
the retention system. Figure 1A shows that pulse-labeled BiP was
secreted from the erd2 strain as efficiently as from the
YFGR strain.
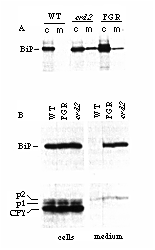
Figure 1. erd2 Cells Secrete BiP
(A) Cells were labeled with 35SO4 for 20
min and chased for 40 min. BiP was then immunoprecipitated from
cells (c) and media (m). Strains analyzed were wild-type (WT),
an isogenic strain whose BiP gene has been modified to delete
the HDEL sequence (FGR), and an erd2 mutant (erd2).
The band slightly larger than BiP in the FGR cell sample is unprocessed
pre-BiP, which tends to accumulate when expression levels are
high (Rose et al., 1989).
(B) The same strains were analyzed by immunoblotting of proteins
from cells and media with anti-BiP (upper panel) or anti-CPY (lower
panel); p1 and p2 refer to the ER and Golgi forms of CPY, respectively.
The autoradiogram of the anti-BiP medium samples was exposed for
5 times as long as the cell samples, for clarity.
Secretion of BiP from the erd2 strain was confirmed by immunoblotting of cells and media (Figure 1B): the erd2 and FGR strains released equivalent amounts of BiP into the medium, whereas the wild type strain did not. Nevertheless, cells of all three strains contained similar levels of the protein. As we have shown previously, the loss of BiP from the FGR strain is compensated for by an increase in its rate of synthesis (Figure 1A). Although it is not obvious in the experiment shown in Figure 1A, other experiments have shown a similar induction of BiP synthesis in erd2 strains.
Also shown in Figure 1B is an immunoblot of the vacuolar enzyme carboxypeptidase Y (CPY). Three forms of the CPY glycoprotein can be identified: the mature vacuolar form, the core-glycosylated ER form of the proenzyme (p1), and the more extensively modified p2 form, which is found in the Golgi (Stevens et al., 1982). Small amounts of the p2 form fail to reach the vacuole and are secreted into the medium. In contrast to erd1 mutants (Hardwick et al., 1990), erd2 cells showed no obvious abnormality in the glycosylation or targeting of CPY (Figure 1B). Thus, the erd2 defect appears specific to the ER retention system.
Cloning of ERD2
A plasmid that complemented the erd2 phenotype was isolated
from a genomic library. Subclones of the 10 kb insert were tested,
and the activity was localized on a 3.3 kb HindIII fragment that
was then sequenced (Figure 2). The sequence contains two open
reading frames. One is present on the strand complementary to
that shown in Figure 2, between bases 2496 and 2960; it potentially
encodes a 155 amino acid protein, but fragments containing this
open reading frame were unable to complement erd2 mutations.
The second open reading frame encodes 219 amino acids, interrupted
by a single intron close to the N terminus with perfect consensus
splice donor, branchpoint, and acceptor sequences (Figure 2; Vijayraghavan
et al., 1986). S1 nuclease mapping confirmed that the region was
transcribed and that splicing occurred in the expected position
(data not shown). An intronless version of this open reading frame,
prepared by oligonucleotidedirected mutagenesis and inserted into
an expression vector (see Experimental Procedures), was sufficient
both to complement an erd2 mutation and to provide other
ERD2-associated functions (see below).

Figure 2 Sequence of the ERD2 Gene
The consensus splice donor, branchpoint, and acceptor sequences
are indicated by double underlining. Dotted lines mark two related
15 amino acid sequences near the N terminus of the protein. Amino
acid changes caused by the mutations listed in Table 1 are shown
in boldface above the wild-type sequence.
To confirm that this open reading frame corresponds to the
gene that is mutated in the erd2 strains, we isolated the
corresponding gene from four independently isolated erd2
alleles using polymerase chain reaction (PCR) amplification. The
PCR products were then cloned and sequenced. Individual clones
frequently contained two or more base changes, but comparison
of several independent clones from each mutant revealed that in
each case only one change was consistently observed, and in three
of the four genes this could be checked by digestion of the uncloned
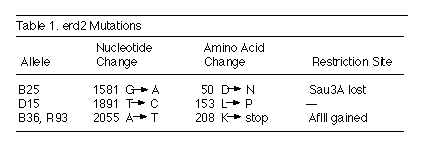
PCR products with restriction enzymes (Table 1). Two of the mutants,
isolated in different genetic backgrounds, had the same base change
that generates a termination codon 12 amino acids from the C terminus.
The other two had single amino acid changes elsewhere in the protein
(Table 1, Figure 2). Thus, all the erd2 mutant strains
analyzed carry mutations in the gene that we have cloned. We therefore
refer to this gene as ERD2.
ERD2 Encodes an Integral Membrane Protein
The predicted ERD2 protein sequence contains no cysteine
residues, nor any potential N-glycosylation sites. Data base searches
revealed no significant homology to other known proteins, but
there is a striking similarity between residues 26-40 and 46-60
(Figure 2), which suggests an ancient intragenic duplication.
A notable feature of the protein is its hydrophobicity, which can be seen from the hydropathy plot in Figure 3. There is no obvious N-terminal signal sequence, but there is a stretch of 17 uncharged amino acids between residues 66 and 82, and hydropathy analysis suggests the presence of several (2-4) transmembrane segments. We therefore sought evidence that the ERD2 protein is membrane associated.
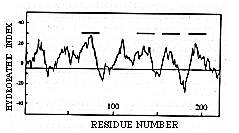
Figure 3. Hydropathy Plot of the Predicted ERD2 Protein The algorithm of Kyte and Doolittle (1982) was used, with a window of 11 amino acids. Hydrophobic portions are above the horizontal line. Bars above the plot indicate potential transmembrane segments.
To facilitate detection of the protein, we tagged the C terminus of the open reading frame with a sequence encoding a 10 amino acid epitope from the human c-myc gene, which is recognized by the monoclonal antibody 9E10 (Evan et al., 1985; Munro and Pelham, 1986). The tagged gene remained functional, as judged from its ability to complement an erd2 mutation (not shown, but see Figure 5 for an example of its activity). Immunoblotting of proteins from a strain expressing this gene revealed a 9E10-reactive band at the expected position for a 26 kd protein, which was absent from a strain lacking the tagged protein (Figure 4A).
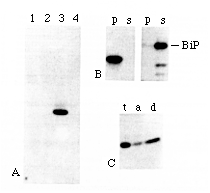
Figure 4. Detection and Membrane Association
of the ERD2 Protein
(A) Total protein from a wild-type strain (lanes 2 and 4) and
from a strain lacking the chromosomal ERD2 gene but carrying
a multicopy plasmid (JS209) that expresses ERD2 protein
tagged with the 9E10 epitope (lanes 1 and 3) was analyzed by immunoblot
using the 9E10 monoclonal antibody. Samples in lanes 1 and 2 were
boiled in SDS sample buffer before gel electrophoresis. The band
in lane 3 had the mobility expected for a 26 kd protein, as judged
from markers run on the same gel.
(B) Membrane fractions enriched for ER were prepared from the
strain expressing tagged ERD2, extracted with sodium carbonate
(pH 11.5), and the membranes then pelleted by centrifugation.
Pellet (p) and supernatant (s) fractions were immunoblotted with
9E10 antibody (lefthand panel) or with anti-BiP (righthand panel).
Note that ERD2 protein remained in the pellet, while BiP
was extracted.
(C) Proteins from the same strain were partitioned between an
aqueous phase and the detergent Triton X-114, and samples of the
total extract (t), the aqueous phase (a), and the detergent phase
(d) were blotted and probed with the 9E10 antibody. A substantial
proportion of the tagged ERD2 protein was recovered in
the detergent phase.
The tagged ERD2 protein sedimented with membranes during centrifugation. It could not be extracted from microsomes with sodium carbonate at pH 11.5, a treatment that is considered to remove all nonintegral proteins from membranes (Fujiki et al., 1982) and that resulted in efficient release of BiP (Figure 4B). Furthermore, a substantial portion of the ERD2 protein partitioned into the detergent phase when cell extracts were extracted with Triton X-114 (Figure 4C). Finally, boiling of cell extracts in SDS prior to gel analysis resulted in a failure of the protein to enter the gel, a commonly observed phenomenon with membrane proteins (Figure 4A, lane 1). Together, these results strongly suggest that the ERD2 gene product is an integral membrane protein.
In preliminary studies, we have used indirect immunofluorescence to locate the tagged protein within cells. The staining observed was punctate, and associated with neither nucleus nor vacuole. It was similar to that obtained with antibodies to the Golgi-associated YPT1 protein (Segev et al., 1988) and distinct from the ER pattern seen with anti-HDEL antibodies (Hardwick et al., 1990). Thus, although caution is required in interpreting the distribution of a protein that has been modified by tagging, it seems likely that much of the ERD2 protein is normally present in a post-ER, Golgi-like compartment. Further analysis will be required to identify its precise location.
ERD2 Expression Level Regulates the Capacity of the HDEL
Retention System
To investigate the role of ERD2, we studied the effect
of its overexpression on the retention of a pro-a
factor-HDEL fusion protein (this protein also carries the c-myc
epitope, to allow specific immunoprecipitation). This fusion protein
has previously been used to demonstrate the recycling pathway
in yeast cells (Dean and Pelham, 1990). It is normally efficiently
exported from the ER, and has two advantages for our purposes:
it is a small glycoprotein, which allows even minor Golgi modifications
to be detected by changes in its electrophoretic mobility, and
when it reaches the later part of the Golgi it is proteolytically
processed to the 13 amino acid a factor (Julius et al., 1984);
thus, the modification state of pro-a
factor retained in the ER can be examined without interference
from secreted molecules.
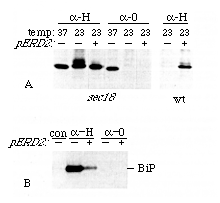
Figure 5. ERD2 Overexpression Increases
the Efficiency and Capacity of the Retention System
(A) sec18 and sec+ (wt) cells expressing
pro-a factor
fusion proteins tagged with HDEL (a-H) or not (a-0), and carrying a multicopy ERD2 plasmid where
indicated, were pulse-labeled for 10 min and chased for 10 min,
and intact pro-a
factor was isolated by immunoprecipitation. Labeling was carried
out either at the sec18 permissive temperature (23°C)
or at the nonpermissive temperature (37°C). Note that the
main band visible is the ER-modified form of the fusion protein;
the pulse-labeled protein in the band above this has been subjected
to Golgi modification, which does not occur in sec18 cells
at 37°C. Overexpression of ERD2 reduces this modification
in sec18 cells and reduces proteolytic processing of the
protein in wild-type cells.
(B) The BiP content of the medium in various cultures was revealed
by immunoblotting. Cells were sec+ and expressed
the a-H construct,
the a-0 construct,
or neither (con). Overexpression of ERD2 reduced the secretion
of BiP that was induced by the a-H construct.
All experiments used a high copy plasmid carrying the normal ERD2
gene (plasmid PER220).
Figure 5A shows the results obtained when sec18 strains expressing pro-a factor fusion proteins were labeled for 10 min, chased for 10 min, and then analyzed by immunoprecipitation and gel electrophoresis. Cells carrying the sec18-1 mutation have a temperature-sensitive lesion in vesicular transport between ER and Golgi; thus at the nonpermissive temperature (37° C), fusion proteins that either contained HDEL (a-H) or lacked it (a-0) were retained in the ER. At the permissive temperature (23°C) the a-0 construct was rapidly lost from the ER, while the a-H protein was retained. Some of the retained protein had been exposed to the Golgi a(1-6)mannosyl transferase, and showed a characteristically reduced mobility. Cells containing ERD2 on a multicopy plasmid also specifically retained the a-H protein, but there was considerably less of the Golgi modified form. This result is consistent with the idea that high levels of ERD2 either slow the exit of HDEL proteins from the ER or promote their retrieval from a compartment prior to the one that contains a(1-6)mannosyl transferase.
In a sec+ strain, high level expression of the a-H construct resulted in its secretion (Figure 5A), a phenomenon that reflects the saturation of the HDEL retention system (Dean and Pelham, 1990). Overexpression of ERD2, however, allowed retention to occur, suggesting that the capacity of the system had been increased. This interpretation was confirmed by examination of BiP secretion from the same strains. Immunoblots of the culture medium (Figure 5B) showed that, whereas the parental strain or one expressing a-0 did not secrete significant quantities of BiP, the strain expressing a-H did, as expected if the retention system is saturated. BiP secretion was reduced when ERD2 was overexpressed, again implying an increase in the capacity of the HDEL system.
As an additional test of the relationship between ERD2 levels and the retention capacity of cells, we measured the efficiency of secretion of an HDEL-tagged invertase fusion protein in strains engineered to contain different levels of ERD2. In the wild-type strain, secretion of the HDEL protein was clearly reduced relative to a control protein that lacked HDEL, but some secretion was observed (Table 2). In a strain carrying ERD2 on a multicopy vector, retention of the HDEL protein was improved. Conversely, in a strain that had the chromosomal copy of ERD2 deleted and carried only a cDNA copy of the gene on a centromere-containing expression vector, the HDEL protein was secreted almost as efficiently as the control. These cells must contain some ERD2 protein because it is required for their growth (see below). However, the mere presence of the wild-type protein is evidently not sufficient for the retrieval system to function effectively.

We conclude from these experiments that the capacity of the HDEL retention system is primarily determined by the level of ERD2 protein. Overexpression of ERD2 increases the efficiency of retention, while mutation of ERD2, or low expression of the wild-type protein, severely reduces the ability of cells to retrieve ER proteins from the Golgi.
ERD2 Overexpression Suppresses the erd Phenotype
of Other Mutants
During our search for mutants with an erd phenotype, we screened
a variety of sec mutants that are known to affect vesicular traffic
between ER and Golgi (sec12, sec13, sec16, sec17, sec18, sec19,
sec20, sec21, sec22, sec23 ). These mutants are temperature-sensitive
for growth, and fail to export proteins from the ER at the nonpermissive
temperature (Novick et al., 1980, 1981). Our expectation was that
some of them might show a partial defect at the permissive temperature,
and that this might interfere with the recycling of ER proteins
from the Golgi to the ER.
Of the mutants tested, four (sec17,sec18, sec20, and sec22 ) showed detectable secretion of HDEL-containing proteins at the permissive temperature, although all had a weaker phenotype than erd2 mutants, and the sec18 signal was very low (Figure 6). These four sec genes represent a specific class, in that they all accumulate pre-Golgi transport vesicles when incubated at the nonpermissive temperature and have elevated levels of these vesicles at the permissive temperature (Kaiser and Schekman, 1990). Mutants that fail to generate transport vesicles did not have an erd phenotype. This suggests that some component necessary for recycling is sequestered in transport vesicles and becomes limiting when they accumulate, slowing the retrieval process and allowing ER proteins to escape.
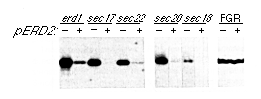
Figure 6. ERD2 Suppresses the erd
Phenotype of Other Mutants
Mutant strains were transformed with an ERD2-containing
plasmid or with a control plasmid, and samples of medium from
the various cultures were analyzed for their BiP content by immunoblotting.
For this experiment, a multicopy plasmid (JS209) expressing a
tagged, intronless copy of the ERD2 gene was used, although
similar results were obtained with the normal gene. The erd1
strain carries a deletion of the ERD1 gene. All sec
mutants, which are temperature-sensitive, were grown at the permissive
temperature (23°C). The FGR strain has its chromosomal BiP
gene modified to remove the HDEL coding sequence; note that ERD2
had no effect on the secretion of this modified BiP.
Figure 6 shows that overexpression of ERD2 prevented the secretion of BiP from all four of the sec mutants, suggesting that it is the limiting component of the recycling system that is present in transport vesicles. ERD2 did not, however, overcome the temperature-sensitive phenotype of these mutants (nor of any of the other sec mutants tested), nor did it prevent the secretion of BiP molecules that lacked the HDEL signal from the YFGR strain (Figure 6). Thus, the effects of ERD2 overexpression are restricted to the HDEL retrieval system the normal process of secretion is unaffected.
ERD2 also suppressed the secretion of BiP from an erd1 deletion mutant (Figure 6). This mutant has a defect that prevents the addition of "outer-chain" mannose residues to glycoproteins in later Golgi compartments, although the early part of the Golgi appears to function normally (Hardwick et al., 1990). Examination of glycoproteins in the suppressed strain showed that ERD2 did not correct the glycosylation defect (not shown); its action can be most easily explained by an increase in the efficiency of retrieval of ER proteins from the early, nondefective Golgi compartments. This is consistent with our previous observation that the extent of Golgi modification of a pro-a factor-HDEL fusion protein is reduced by overexpression of ERD2 (Figure 5).
ERD2 Is Essential for Growth
To investigate the effects of a complete absence of the ERD2
protein, we deleted from the cloned gene an NcoI fragment that
includes all but the last 59 codons of the open reading frame
and some 700 bp of 5' flanking sequence (bases 600 to
1915 in Figure 2), and introduced this deletion into one chromosome
of a diploid strain by the two-step procedure of Boeke et al.
(1984). Deletion of the fragment was confirmed by Southern blot
analysis (not shown). Sporulation of this strain revealed a recessive
lethal mutation. Ten tetrads showed a segregation pattern of two
viable and two nonviable spores; the latter germinated and divided
three times on average, but then ceased to grow.
Confirmation that ERD2 protein is essential for growth was provided by an additional experiment. The ERD2 open reading frame was fused to the inducible GAL1 promoter and integrated at the URA3 locus of the diploid strain that carried the erd2 deletion. Sporulation of this strain gave rise to some haploid cells whose only copy of ERD2 was under GAL1 control. These cells were grown in medium containing galactose to activate transcription from the GAL1 promoter (Johnston and Davis, 1984); under these conditions, they divided as rapidly as wild-type cells. However, when the GAL1 promoter was inactivated by transferring the cells to glucose-containing medium, the strain lacking the normal ERD2 gene gradually ceased to grow (Figure 7). The cells remained quiescent but did not die: 2 days after they had ceased to grow, 80% of the cells were still capable of forming colonies on galactose-containing plates. Thus, growth can be reversibly halted by depletion of the ERD2 protein.
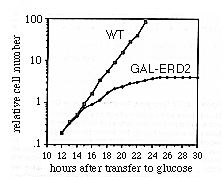
Figure 7 ERD2 Expression Is Essential
for Growth
A strain was constructed that lacked the normal ERD2 gene
but carried an integrated copy of the ERD2 coding sequence
under the control of the GAL1 promoter (see Experimental
Procedures). This strain (solid symbols) and an ERD2+
control (open symbols) were maintained in galactose-containing
medium, then transferred to glucose medium and their subsequent
growth monitored. The cultures were diluted at 17 hr to maintain
logarithmic growth conditions.
Protein Traffic through the Golgi Is Defective in Cells
Lacking ERD2
The growth defect of cells lacking ERD2 was unexpected,
because previous studies had suggested that the HDEL retention
system is dispensable (Hardwick et al., 1990), and indeed the
growth-arrested cells had normal levels of BiP (not shown). It
seemed therefore that ERD2 must perform a second function;
to identify this function, we examined the properties of ERD2depleted
cells.
Figure 8A shows an electron micrograph of a cell containing the GAL1-ERD2 fusion gene that had been grown on glucose for 24 hr. For comparison, wild-type cells (Figure 8B) and sec20 and sec7 cells that had been incubated for 3 hr at the nonpermissive temperature, which induces the accumulation of ER and transport vesicles (sec20; Figure 8C) or Golgi like structures (sec7 ; Figure 8D) (Novick et al., 1980; Kaiser and Schekman, 1990), are also shown. The erd2-deficient strain accumulated higher than normal amounts of intracellular membranes. This phenotype was detected in some cells as early as 14 hr after transfer to glucose; after longer times it was apparent in most cells. The membranes seemed more fragmented than the ER that accumulates in mutants such as sec20 , which typically show much longer continuous ribbons of ER than are seen in wild-type cells. There was no significant accumulation of vesicles, nor of the characteristically bloated Golgi structures seen in the sec7 mutant. However, since the distended cisternae seen in sec7 cells are not present in wild-type cells, which nevertheless have Golgi compartments, it remains possible that some of the membranes in theerd2 -- depleted cells are Golgi derived. All the mutants accumuleted lipid droplets, visible as white areas in the micrographs.
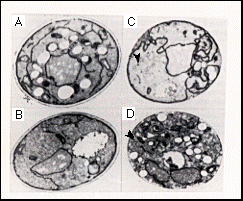
Figure 8. Accumulation of Membranes in ERD2
-Depleted Cells
Electron micrographs of permanganate-fixed cells are shown. Total
width of each panel corresponds to 3 µm.
(A) A cell carrying the GAL1-ERD2 fusion gene, 24
hr after transfer to glucose; note abundant ER-like membranes.
Clear white areas are lipid droplets.
(B) An ERD2+ control, treated as in (A).
(C) A sec20 cell after 3 hr at 37°C; note accumulation
of continuous strands of ER, together with vesicles (arrowhead).
(D) A sec7 cell after 3 hr at 37°C; note Golgi-like
structures with poorly stained lumen (arrowhead).
These results are consistent with a block in the secretory pathway in erd2-depleted cells, but the phenotype appeared different from that of any known sec mutant. To identify the step that becomes rate limiting, we examined the processing of CPY in the absence of ERD2. Cells carrying the GAL1-ERD2 fusion gene were grown for 15 hr in glucose medium and their CPY content was analyzed by immunoblotting (Figure 9A). At 15 hr, growth of the cells was just beginning to slow down (Figure 7); similar results were obtained after 24 hr, when growth had ceased entirely. Relative to the control cells, there was no change in the level of the p1 ER form of CPY, but there was a marked increase in the amount of the p2 Golgi form, suggesting that passage of CPY through the Golgi was inhibited.
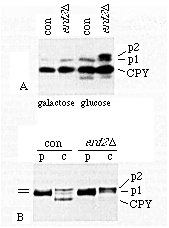
Figure 9. ERD2-Deficient Cells Accumulate
the Golgi Form of CPY
(A) Cells carrying the GAL1-ERD2 fusion gene (ERD2D) and ERD2+
controls (con) were grown in galactose medium, or transferred
to glucose medium for 15 hr, washed, and their CPY content analyzed
by immunoblotting. Similar results were obtained after 24 hr in
glucose.
(B) Control and ERD2-deficient cells, after 15 hr growth
in glucose, were pulse-labeled for 10 min (p) and chased for 30
min (c). Intracellular CPY was then isolated by immunoprecipitation.
The Golgi precursor of CPY (p2) accumulated in the mutant cells
in both experiments, while the ER form (p1) appeared normal.
The transport defect was confirmed by pulse-chase analysis (Figure 9B). Synthesis, translocation into the ER, and conversion of the ER form of CPY to the Golgi form occurred with normal kinetics in the arrested cells, but vacuolar processing was substantially delayed or inhibited (Figure 9B). In some experiments the accumulated p2 form in the mutant cells appeared to have a very slightly increased mobility relative to the control cells, suggesting that its modification state might be different. However, at least some of this precursor was precipitable with antibodies that recognize a(1-3)mannose (not shown), as is normal p2 (Franzusoff and Sckekman, 1989). Since these residues are added in a late Golgi compartment, transport through most of the Golgi must have occurred, though perhaps more slowly than usual. The p2 CPY remained associated with the cells and therefore was not simply mistargeted to the cell surface. It also seems unlikely that it was transported to the vacuole but failed to be processed, since the cells still contained high levels of mature CPY (Figure 9A) and presumably also other vacuolar proteases.
These results imply that ERD2 function is required, directly or indirectly, for normal transport of CPY (and presumably other proteins) through the Golgi apparatus. Since there is an accumulation of intracellular membranes in the absence of ERD2, it would appear that the defect is due to inefficient vesicular transport.
Discussion
Our genetic analysis of the retention of HDEL proteins in yeast has so far identified only two genes that are required for this purpose. The ERD1 gene is not required when cells are grown in certain minimal media (Hardwick et al., 1990), but the ERD2 gene appears to be needed under all conditions. The ERD2 product is therefore likely to be closely involved in the retention mechanism. The results reported here indicate that it is a 26 kd integral membrane protein that possibly contains multiple membrane-spanning segments. Preliminary immunofluorescence studies suggest that much of it is found in a postER, Golgi-like compartment.
The Role of ERD2 in Retention of Luminal ER Proteins
A striking observation is the correlation between the level of
ERD2 expression and the capacity of the HDEL retention
system. Thus, while mutation or underexpression of the gene causes
secretion of ER proteins such as BiP, overexpression improves
their retention in wild-type cells without affecting the secretion
of other proteins. In particular, when the HDEL system is saturated
by the abundant synthesis of an HDEL-bearing fusion protein, high
levels of ERD2 alleviate the problem and reduce secretion
of both the fusion protein and endogenous ER proteins. This ability
to increase capacity suggests that ERD2 controls the concentration
of the HDEL receptor in the sorting compartment; the simplest
interpretation is that ERD2 encodes the receptor.
Although this argument is suggestive, it is not conclusive. However, in the accompanying paper it is shown that replacement of the ERD2 gene with the equivalent gene from Kluyveromyces lactis (another budding yeast) changes the signal specificity of the retention system (Lewis et al., 1990). Thus ERD2 determines both the capacity and specificity of the retention system, providing strong evidence that it is indeed the receptor.
If so, the ability of ERD2 to suppress the weak erd phenotype of the sec mutants that accumulate pre-Golgi transport vesicles is readily explained. Presumably, HDEL receptor is present in the accumulated vesicles and is correspondingly depleted from the sorting compartment. Providing more receptor would compensate for this effect. It could also encourage retrieval from early compartments on the secretory pathway, thus accounting for the suppression of BiP secretion from erd1 mutants, which are defective only in the later regions of the Golgi (Hardwick et al., 1990).
The Role olf ERD2 in Secretion
The fact that ERD2 is essential for growth suggests that
it has a second function, distinct from its role in the targeting
of soluble ER proteins. This function seems to be required for
the correct operation of the secretory pathway. Specifically,
transport of CPY through the Golgi is impaired when the ERD2
protein is depleted from cells, and intracellular membranes accumulate.
It seems very unlikely that this defect is merely a consequence of the failure to retain resident soluble proteins in the ER. Retention of BiP by the HDEL system is certainly not essential for growth, because a strain whose only BiP gene lacks the sequences encoding HDEL grows normally. Viable erd2 mutants secrete BiP at the same rate as this strain, and thus show no detectable HDEL-dependent retention (Figure 1). Moreover, any residual receptor activity can be saturated by overexpression of a pro-a factor-HDEL fusion protein, with no obvious ill effect. Finally, if the growth defect were due to the reduction of the level of some hypothetical luminal ER protein below a critical threshold, one might expect to find a defect in ER function. In contrast, we find that translocation of CPY into the ER, its glycosylation, and transport out of the ER all appear normal in the absence of ERD2; rather, it is traffic through the Golgi that is affected, at least initially.
What might be the role of ERD2 in Golgi transport? One possibility is that this small protein has two quite separate functions, one being to bind HDEL proteins and the other being essential for the maintenance of Golgi structure or physiology. A more attractive possibility is that the two functions are related. If the ERD2 protein is the HDEL receptor, it follows that it must cycle between Golgi and ER. It seems likely to be a major component of the vesicles that return from the Golgi, perhaps the most abundant membrane protein in them. It would not be surprising if it were required for such vesicles to form. For example, by clustering in the membrane and associating with a suitable coat protein, ERD2 protein could help to form a structure analogous to a coated pit. If it plays such a role, then in its absence no proteins, either soluble or transmembrane, could return from the Golgi to the ER. It is not clear whether any ER membrane proteins normally escape to the Golgi and require retrieval, but recycling of the SEC12 protein, and perhaps other membrane proteins involved in vesicular traffic, has been suggested (Nakano et al., 1989). ERD2 might be necessary to maintain the supply of such components in the compartments from which vesicles bud. Alternatively, their accumulation in an inappropriate compartment might cause mistargeting of transport vesicles. In either case, a failure to retrieve these membrane proteins could be sufficient to reduce the flow of the secretory pathway to a trickle.
Experimental Procedures
The strains used in Figure 1 (and part of Table 2, as indicated)
were derivatives of SEY2102 (MATa
ura3-52 leu2-3,-112 his4-519 suc2-d9). Other constructed
strains (except for sec strains) were derived from SEY6210 (MATa ura3-52 his3-d200 leu2-3,-112 trp1-d901
Iys-801 suc2d9) or, for the erd2 deletion strains,
from a diploid obtained by crossing SEY6210 with SEY6211 (MATa
ura352 his3-d200 leu2-3,-112 trpl-d901suc2-d9 ade2 101). The
sec18 strain used in Figure 5 is ND1811 (MATa secl8-1 ura3-52
suc2d9) (Dean and Pelham, 1990). The sec7 strain used for electron
microscopy was N D72 (sec7-1 ura352 trp1-d901 his3-d200 leu2-3,-112
suc2-d9). Other sec mutant strains were kindly provided by
Chris Kaiser: RSY263 (secl2-4), RSY265 (secl3-1),
RSY267 (secl6-2), RSY269 (secl7-1), RSY271 (secl8-1),
RSY273 (sec19-1), RSY275 (sec20-1), RSY277 (sec21
1), RSY279 (sec22-3), RSY281 (sec23-1) (Kaiser
and Schekman, 1990).
ERD2 Plasmids
The ERD2 gene was isolated by complementation, using a
library constructed in a centromere vector (Sengstag and Hinnen,
1987). An intronless version of the open reading frame was prepared
by PCR (Higuchi, 1989), using a primer encoding the first 12 amino
acids of the protein. A PstI site was introduced immediately after
the last codon by similar means and was then used to fuse the
open reading frame to DNA encoding the epitope (EQKLISEEDLN) recognized
by the 9E10 monoclonal antibody, generating a tagged version of
the gene. The structure of the PCR-generated constructs was verified
by sequencing. Mutant erd2 alleles were isolated from genomic
DNA by PCR, using primers that flank the coding sequence.
Four expression plasmids were used for the experiments described in this paper. Plasmid PER220 contains the HindIII fragment whose sequence is shown in Figure 2, inserted into the LEU2-containing 2µm vector ZUC13. JS209 contains a tagged, intronless copy of the ERD2 coding sequence fused to the triose phosphate isomerase (TPI) promoter, on a URA3 containing 2µm vector JS214 contains an intronless but untagged gene, also fused to the TPI promoter, on a vector with TRP1, ARS1, and CEN3. HP208 contains the untagged, intronless gene fused to the GAL1 promoter, on a vector (YIP56X) that allows integration at the ura3-52 locus (Pelham et al., 1988).
To delete the chromosomal ERD2 gene, the Ncol fragment that extends from nucleotide 597 to 1912 was deleted from the HindIII fragment (Figure 2) and the deleted fragment inserted into the URA3-containing integration vector YIP56. This was linearized at the single remaining KpnI site (nucleotide 2682) and integrated at one copy of the ERD2 locus in a diploid strain (generated by crossing SEY6210 and SEY6211). After 5 fluoro-orotic acid selection (Boeke et al., 1984), a strain carrying one deleted copy of the gene was identified by Southern blot analysis. This was transformed, separately, with JS209, JS214, and HP208. The resulting strains were sporulated, and spores lacking the chromosomal gene identified by Southern blot analysis (and, in the case of HP208, by their dependence on galactose medium for growth). The resulting strains were designated D209, D214, and D208, respectively. Other ERD2 expression plasmids were introduced into the deletion background by transfection of strain D209 followed by 5-fluoroorotic acid selection to eliminate JS209.
Plasmids containing the invertase and prepro-a factor fusion genes have been described elsewhere (Pelham et al., 1988; Dean and Pelham, 1990). The a factor constructs include the coding sequence for the 9E10 epitope, allowing specific immunoprecipitation of the expressed protein with monoclonal antibody 9E10 without interference from endogenous u factor. Invertase-BiP fusion proteins were expressed from the PRC1 promoter, prepro-a factor constructs from the TPI promoter; both were integrated at URA3, and the number of integrated copies was checked either by Southern blot analysis or by quantitative invertase assays.
Detection of Tagged ERD2 Protein
Crude ER fractions were prepared using a sucrose step gradient
as described by Dean and Pelham (1990). Samples were diluted 10-fold
in 0.1 M sodium carbonate (pH 11.5), incubated for 30 min at 0°C,
and then centrifuged for 1 hr at 245,000 x g in a Beckman TLA-100.2
rotor, essentially as described by Fujiki et al. (1982). Triton
X-114 extraction of whole spheroplasts was performed as described
by Lewis et al. (1985),
Other Techniques
Immunoblotting of cells and media, pulse-labeling with 35SO4
and immunoprecipitation, invertase assays, and electron microscopy
were performed as previously described (Dean and Pelham, 1990;
Hardwick et al., 1990; Pelham et al., 1988).
Acknowledgments
K. G. Hardwick and J. C. Semenza made equal contributions to the work described in this paper We thank Deborah Sweet for the data shown in Table 2, Mark Rose, Tom Stevens, and Gerard Evan for antibodies, Albert Hinnen for the genomic library, Chris Kaiser for yeast strains, Mike Lewis for plasmids, Nichol Thomson and Douglas Kershaw for help with electron microscopy, and Sean Munro, Mark Bretscher, and Barbara Pearse for comments on the manuscript. N. D. was supported by a fellowship from the American Cancer Society, and J. C. S. by the Roche Research Foundation.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received May 10, 1990.
References
Boeke, J. D., Lacroute, E, and Fink, G. R. (1984). A positive selection for mutants lacking orotidine-5'phosphate decarboxylase activity in yeast: 5 fluoro-orotic acid resistance. Mol. Gen. Genet. 197, 345346.
Dean, N., and Pelham, H. R. B. (1990). Recycling of proteins from the Golgi compartment to the ER in yeast. J. Cell Biol., in press.
Evan, G. l., Lewis, G. K., Ramsay, G., and Bishop, J. M. (1985). Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 5, 3610-3616.
Franzusoff, A., and Schekman, R. (1989). Functional compartments of the yeast Golgi apparatus are defined by the sec7 mutation. EMBO J. 8, 2695 2702.
Fujiki, Y, Hubbard, A., Fowler, S., and Lazarow, P B. (1982). Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93, 97-102.
Hardwick, K. G., Lewis, M. J., Semenza, J., Dean, N., and Pelham, H. R. B. (1990). ERD1, a yeast gene required for the retention of luminal endoplasmic reticulum proteins, affects glycoprotein processing in the Golgi apparatus. EMBO J. 9, 623-630.
Higuchi, R. (1989). Using PCR to engineer DNA. In PCR Technology- Principles and Applications for DNA Amplification, H. A. Erlich, ed. (New York: Stockton Press), pp. 61-70.
Johnston, M., and Davis, R. W. (1984). Sequences that regulate the divergent GALl-GAL10 promoter in Saccharomyces cerevisiae. Mol. Cell. Biol. 4, 1440-1448.
Julius, D., Schekman, R., and Thorner, J. (1984). Glycosylation and processing of yeast prepro-a factor through the yeast secretory pathway. Cell 36, 309-318.
Kaiser, C. A., and Schekman, R. (1990). Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway Cell 61, 723-733.
Kyte, J., and Doolittle, R. F (1982). A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105-132.
Lewis, M. J., Sweet, D. J., and Pelham, H. R. B. (1990). The ERD2 gene determines the specificity of the luminal ER protein retention system. Cell 61, this issue.
Lewis, V, Green, S. A., Marsh, M., Vihko, P, Helenius, A., and Mellman, l. (1985). Glycoproteins of the lysosomal membrane. J. Cell Biol. 100, 1839 1847
Mazzarella, R. A., Srinivasan, M., Haugejorden, S. M., and Green, M. (1990). ERp72, an abundant luminal endoplasmic reticulum protein, contains three copies of the active site sequences of protein disulfide isomerase. J. Biol. Chem. 265, 1094-1101.
Munro, S., and Pelham, H. R. B. (1986). An hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 46, 291-300.
Munro, S. and Pelham, H. R. B. (1987). A C-terminal signal prevents secretion of luminal ER proteins. Cell 48, 899-907
Nakano, A., Brada, D., and Schekman, R. (1989). A membrane glycoprotein, Sec12p, required for protein transport from the endoplasmic reticulum to the Golgi apparatus in yeast. J. Cell Biol. 107 851-863.
Novick, P, Field, C., and Schekman, R. (1980). Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell 21, 205-215.
Novick, P, Ferro, S., and Schekman, R. (1981). Order of events in the yeast secretory pathway. Cell 25, 461-469.
Palade, G. (1975). Intracellular aspects of the process of protein synthesis. Science 189, 347-358.
Pelham, H. R. B. (1988). Evidence that luminal ER proteins are sorted from secretory proteins in a post-ER compartment. EMBO J. 7,17571762.
Pelham, H. R. B. (1989). Control of protein exit from the endoplasmic reticulum. Annu. Rev. Cell Biol. 5, 1-23.
Pelham, H. R. B., Hardwick, K. G., and Lewis, M. J. (1988). Sorting of soluble ER proteins in yeast. EMBO J. 7, 1757-1762.
Pfeffer, S. R., and Rothman, J. E. (1987). Biosynthetic protein transport and sorting by the endoplasmic reticulum and Golgi. Annu. Rev Biochem. 56, 829-852.
Rose, J. K., and Doms, R. W (1988). Regulation of protein export from the endoplasmic reticulum. Annu. Rev. Cell Biol. 4, 257-288.
Rose, M. D., Misra, L. M., and Vogel, J. P (1989). KAP2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell 57, 1211-1221.
Segev, N., Mulholland, J., and Botstein, D. (1988). The yeast GTPbinding YPT1 protein and a mammalian counterpart are associated with the secretion machinery Cell 52, 915-924.
Sengstag, C., and Hinnen, A. (1987). The sequence of the Saccharomyces cerevisiaa gene PH02 codes for a regulatory protein with unusual aminoacid composition. Nucl. Acids Res. 15, 233-246.
Stevens, T, Esmon, B., and Schekman, R. (1982). Early stages in the yeast secretory pathway are required for transport of carboxypeptidase Y to the vacuole. Cell 30, 439-448.
Vijayraghavan, U., Parker, R., Tamm, J., Iimura, Y, Rossi, J., Abelson, J., and Guthrie, C. (1986). Mutations in conserved intron sequences affect multiple steps in the yeast splicing pathway, particularly assembly of the spliceosome. EMBO J. 5, 1683-1695.
Zagouras, P, and Rose, J. K. (1989). Carboxy-terminal SEKDEL sequences retard but do not retain two secretory proteins in the endoplasmic reticulum. J. Cell Biol. 109, 2633-2640.
GenBank Accession Number
The accession number for the sequence reported in this paper is M34777.
![]()
![]()
©
Copyright 2000 Department of Biology, Davidson College, Davidson,
NC 28036
Send comments, questions, and suggestions to: macampbell@davidson.edu