Focused Reading: "The polymerase..." pp 363-364
Stop at "Antisense..."
"DNA fingerprinting..." 369-370
"Gel electrophoresis..." pp 353-354
WWW Reading: PCR Methods
The first PCR Movie requires that your computer have QT 3.0
The second PCR Movie requires the plugin for Shockwave.
Unless you spent the last few years in a cave, you have heard about the increasing use of "DNA fingerprinting" in court cases. The technology available is so sensitive that unbelievable sources of DNA have been used to convict criminals. In Minnesota for example, DNA was extracted from the back of a postage stamp since some epidermal cells from the tongue had been deposited on the glue when the stamp was licked. My prediction is that the pivotal point in future court cases will be the collection and handling of the evidence. For example, what if some DNA from the crime scene is shown to be the accused? The defense attorney could suggest that the police collected some epidermal cells from the sidewalk (from a visit the day before) at the same time as the blood drops. What do you think of this possibility as a defense? If you want to read more about this area, the library has several books in the area of call numbers 614.1.
There are two standard methods for "DNA fingerprinting": 1) Southern blots and, 2) PCR. We have discussed Southern blots ad nosium in class but have not covered PCR in much detail. Dr. Kary Mullis, the inventor of PCR, was awarded a Nobel Prize in 1993 for his revolutionary innovation. As you know, PCR allows you to amplify a single copy of DNA into millions of copies, provided the DNA has been sequenced because you have to supply DNA polymerase with primers that will specifically hybridize to the target gene and no other DNA. Over the next two weeks, we will use PCR to determine the genotype of every student in class. We are using a hair root as our source of genomic DNA and are looking at a locus called D1S80. D1S80 contains a Variable Number of Tandem Repeat sequence (VNTR). As the term implies, there is a section of DNA that is repeated to varying degrees in each person. As a simplified example, the repeat unit is the two nucleotides (CG). So if we were to sequence this portion of 4 different D1S80 alleles, we might see the following:
1) ATGCCGTATTACGCGCGCGCGCGCGCCTATTAGGTATTAG
2) ATGCCGTATTACGCGCGCGCGCGCGCGCGCGCGCGCCTATTAGGTATTAG
3) ATGCCGTATTACGCGCGCGCCTATTAGGTATTAG
4) ATGCCGTATTACGCGCGCGCGCGCGCGCGCCTATTAGGTATTAG
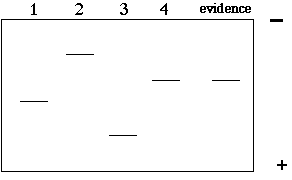
In this example, there are 4 copies of the VNTR with 4 different lengths. If we electrophoresed these 4 segments of DNA on a gel, we would find 4 different bands of different sizes (2>4>1>3). In a criminal case, we might have 4 suspects and one DNA sample from the crime scene. The resulting gel might look like this:
Questions:
1) So, "who done it"?
2) What is wrong with the above gel? Why is this too easy?
Week 1
Now it is time for us to determine our genotypes. You should be forewarned, this is a delicate procedure that does not always work for everyone. In order to process this kind of evidence for a criminal case, a technician usually has a master's degree in Forensic Science, and a few years of "on-the-job training". Nevertheless, even these experts sometimes make mistakes. So do not be discouraged if your sample does not "work", but try to avoid this situation by observing these guide lines:
1) Follow the protocol as carefully as possible.
2) Do not contaminate your hair or DNA with that of others (remember
one cell contains enough
DNA to be amplified).
3) Immediately after the DNA extraction is finished, visually
check to verify that you have
extracted DNA by gently removing the tube from the thermocyler
and flicking the tube
while holding it up to a light and looking very carefully. You
should see a more dense
portion of the solution at the bottom of the tube as it mixes
with the less dense water.
4) The most common mistakes are pipetting errors. Be sure to visually
check to see that
you are transferring about the right volumes and always use clean
tips; when in doubt
get a new one.
5) Be very careful loading the gel. We will have time to practice
this week so that when
you are loading your real sample next week, you will be a pro.
DNA extraction1
1) Pluck a hair so that a large portion of root is removed from your head (yikes!) For most of you, the root will be white/translucent in appearance. People of African heritage will have roots that are dark. Regardless of the color, it will be sticky so you can test it by touching it to the bench top to see if it adheres at all. Check to make sure you got some root and not all shaft.
2) Put the hair into a small microfuge tube with the root at the bottom of the tube. Cut off most of the hair but keep the root (~5mm). Be careful, sometimes the root will jump away when you cut the hair.
3) Incubate the root in 100 µl digestion buffer (which contains 6 µg of proteinase K) for 1 hour at 55° C then 10 minutes at 95° C (what is the purpose of this step?). Use thermocycler program HAIR 1 - lid disabled.
During this waiting period, we will practice loading gels so you will be ready for next week.
PCR Reaction Mixtures
4) When the DNA extraction cools, vortex the tubes for 30 seconds and then set up a new 500 µl microfuge tube by adding the following:
| Reagent | Volume | Final Concentration |
| extracted DNA | 15.0 ul | ~100 ng of DNA |
| reaction mixture | 10.0 ul | see below** |
** the reaction mixture contains the following cocktail:
| Reagent | Volume | Final Concentration |
| H2O | 4.00 ul | |
| 10X PCR buffer (without Mg) | 2.50 ul | 1.5 mM MgCl2 |
| DMSO | 1.25 ul | 5% v/v |
| 20X dNTP's | 1.25 ul | 200 uM each |
| #1 primer | 0.50 ul | 100 ng primer |
| #2 primer | 0.50 ul | 100 ng primer |
| Total Volume | 10.00 ul |
PCR
This locus requires hotstart PCR which means that the Taq DNA polymerase is not added to the PCR mixture until it has been heated to 95° C. The hotstart is necessary because the D1S80 primers have a tendency to anneal to each other rather than the template while the mixture is heating up for the first time, which allows the DNA polymerase to generate "primer dimers". If addition of the DNA polymerase is delayed, then inappropriately annealing primers are denatured as the kinetic energy increases, so no replication occurs until the temperature is lowered later in the procedure, allowing the primers to anneal to the proper portion of the template DNA. DMSO has been included in the reaction mixture to enhance the specificity of the primers.
Step 1: 5 minutes at 95° C (pause during this step for
hot start see below)
Step 2: 1 minute at 95° C
Step 3: 1 minute at 65° C
Step 4: 1 minute at 72° C
Step 5: repeat steps 2 - 4 twenty-nine more times
Step 6: hold at 20° C
5) To initiate hot start PCR, denature the DNA by incubating the tubes for 5 minutes at 95° C (Step 1), maintain the tubes at 95° C while you add 0.4 µl Taq DNA polymerase to each tube. Do not allow the tubes to cool and do not take time to mix the reaction mixture after adding the Taq polymerase.
6) Resume the same PCR program with the heated lid enabled.
7) When the PCR is completed, the tubes are removed and stored at 4° C until next lab meeting.
o >80% of all populations tested are heterozygous
o 28 alleles have been published
o repeat unit is 16 nucleotides long
o if there were zero repeat units, the PCR product would be 142
bp long
o PCR products range from 430 to 814 base pairs long
o 41 repeated units have been observed in the largest allele
o Primer sequences2:
#1 5' GAAACTGGCCTCCAAACACTGCCCGCCG 3'
#2 5' GTCTTGTTGGAGATGCACGTGCCCCTTGC 3'
Footnotes:
1. (Adapted from: PCR Technology by Henry A. Erlich, W. H. Freeman and Co., NY, 1992, pp. 35-37.)
2. Budowle, B., Chakraborty, R., Giusti, A. M., Eisenberg, A. J., and Allen, R. C. (1991) Analysis of the VNTR locus D1S80 by the PCR followed by high-resolution PAGE. American Journal of Human Genetics 48: 137 - 144.
© Copyright 2000 Department of Biology,
Davidson College, Davidson, NC 28036
Send comments, questions, and suggestions to: macampbell@davidson.edu