How to Find and Format Genome Sequences for Whole Genome Analysis
Whole genome projects have the potential to offer a valuable set of genetic and evolutionary information for organisms. In order to undertake large scale whole genome projects, you must obtain and properly format genome sequences so that they may be used in conjunction with analysis and comparison software tools. This webpage provides a step by step tutorial on how to successfully download, format and compare whole proteomes using the software linked to this page (Note: The programs used for this work are available only on Mac systems).
Tools and Programs Used:
DOE Joint Genome Institute Website
Rapid Annotation using Subsystem Technology (RAST) Website
Microsoft Word
FASTA Converter (Download Available)
SubEthaEdit
Terminal
Where to Find Genomes
There are a number of both public and private sources from which you can obtain genome sequences. Examples of these databases are listed and described below:
United States Department of Energy Joint Genome Institute (JGI)
The DOE Joint Genome Institute is a government funded organization created in 1997 to combine intelligence and resources for DNA sequencing. JGI has created a genome database available for public us, as well as a number of software tools useful in genome analysis.
National Microbial Pathogen Data Resource Rapid Annotation using Subsystem Technology (RAST)
RAST is a public online genome analysis tool that provides fully automated annotation services for bacterial and archaeal genomes. While this service is available to the public, users should create a free account in order access data and maintain organization.
How to Download a Whole Genome Sequence
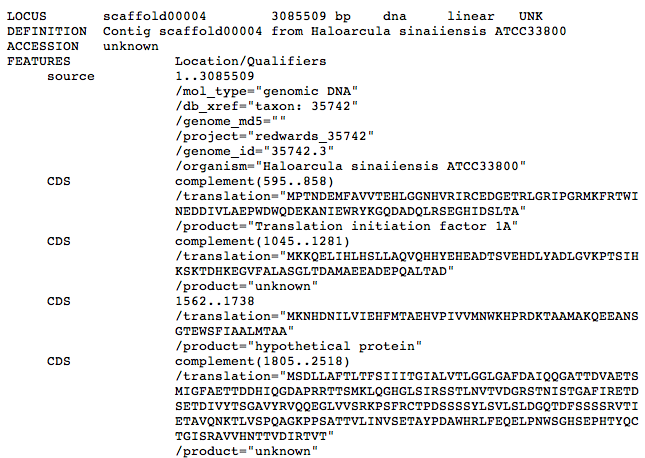
The genome comparison program provided requires access to downloadable files in TextEdit (.txt) format. The program can be used for comparison of proteomes, however all genome sequence files must be in GenBank format. An example of GenBank format is given below.
The original proteome files used in our research were obtained from an external webpage link, the Halophile Sequence Site. The creators of this resource submitted fasta files to RAST and posted the resulting proteome annotations on their webpage.
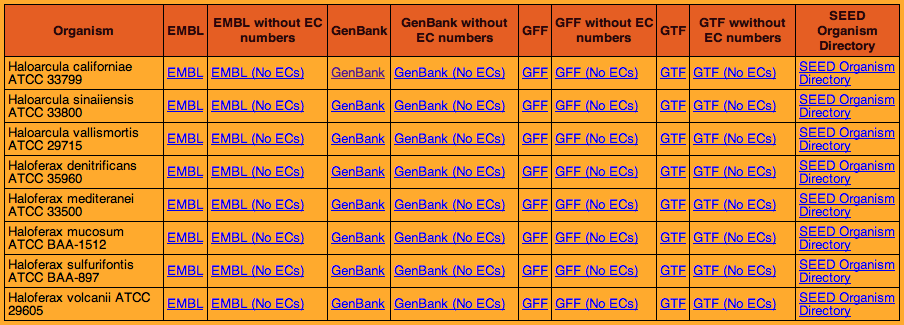
Selecting the GenBank format of the desired proteomes led to the page shown below.
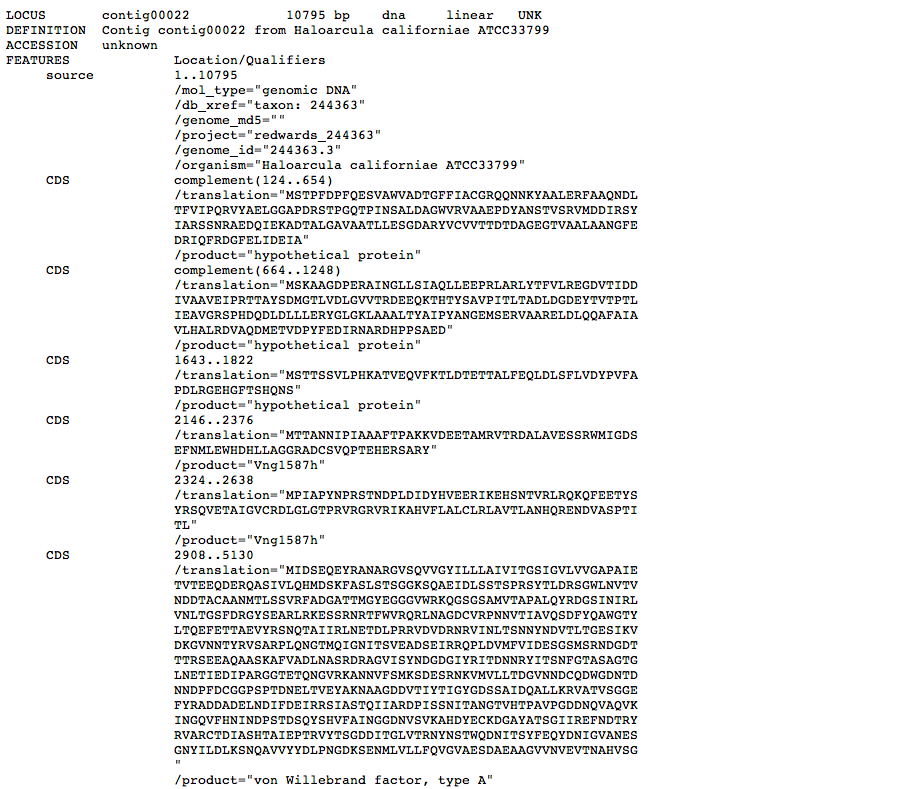
The resulting file text of the entire genome can easily reach upwards of 6 million characters, so the most efficient way of extracting the entire genome data is to use ' Select All ' (Command + A on Mac; or Edit > Select All) and copy the text into Microsoft Word, or a similar word processing program. To minimize file size and complexity, save the file under the desired filename as a TextEdit (.txt) file.
How to Convert GenBank Format to FASTA Format
The file must now be converted to FASTA file format (.fasta) in order to be used with our comparison program.While it is possible to convert sequences into FASTA format manually and individually, it is highly inefficient due to the large number of proteins and characters in the GenBank file. Linked below is a script using BioPerl designed to systematically do this conversion for the entire genome.
First, select the link above. This will download the compressed ZIP file to your desktop or to the location of your choice.
![]()
Then, unzip the file by double-clicking this icon or right-clicking and selecting ' Open '. This will produce the folder pictured below on your desktop once the file has been unzipped.

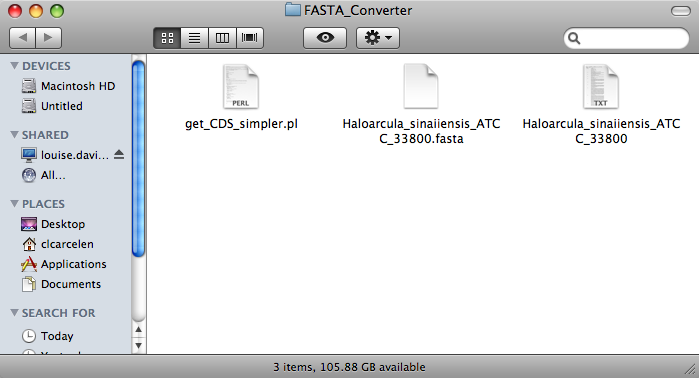
Opening this folder should produce the a window like the one pictured below.
Select the ' get_CDS_simpler.pl ' file.
The file will open in SubEthaEdit, a real-time editing program designed for Mac OS X. You should see the window pictured below.
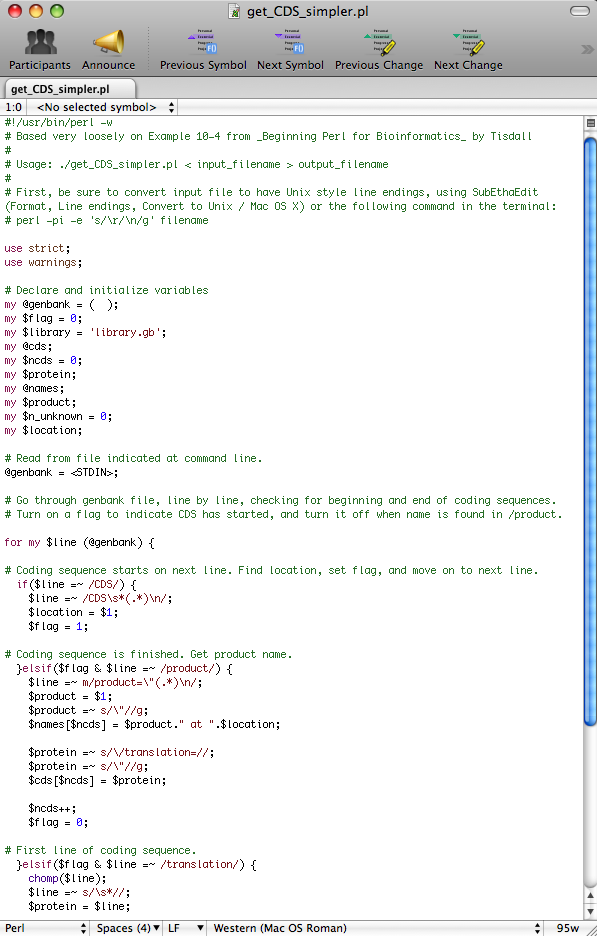
Before running the conversion program, you must ensure that there are no line breaks in the sequences. The presence of line breaks will cause errors in the output. To eliminate any line breaks, open the desired proteome file (which you should have saved earlier as a .txt file). This can be done either by opening SubEthaEdit, selecting ' Open ' and selecting the desired .txt file, or by clicking and dragging the desired file onto the SubEthaEdit icon.
![]()
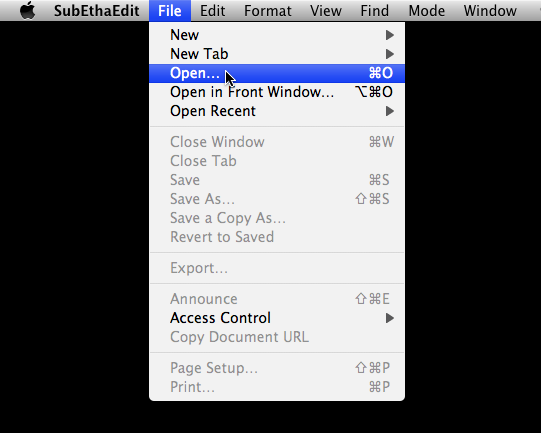
Once the file has opened in SubEthaEdit, a window like the one pictured below should appear.

In the options bar at the top, select ' Format ' > ' Line Endings ' > ' Convert to Unix/Mac OS X Line Endings (LF) '.
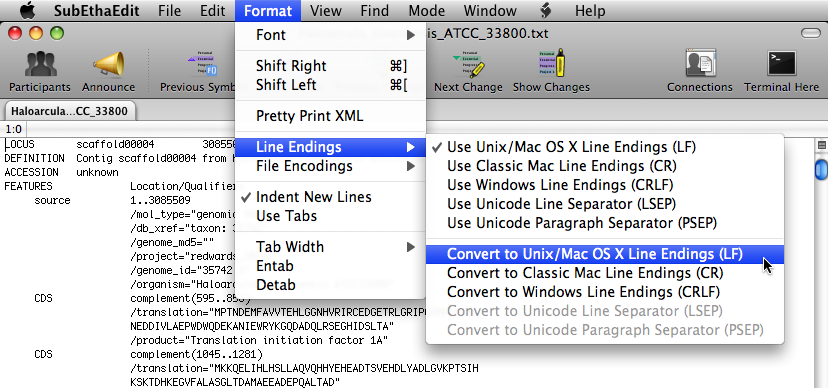
Then, save the file.
In order for the program to work correctly, the file intended for conversion needs to be in the same folder as the program itself. Do so by dragging the file into the FASTA_Converter folder.

To run the program, open the Terminal application. This program is a text-based application that allows you to interact directly with the BSD operating system.
![]()
A window similar to the one shown below should appear on your screen.
Type "cd" followed by a space. (This changes the directory.)
Direct the system by inserting the FASTA_Converter file name. Do so by dragging the file into the Terminal window and the information will appear in the text line in Terminal, as shown below.
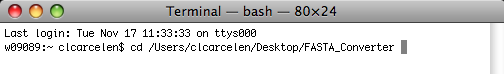
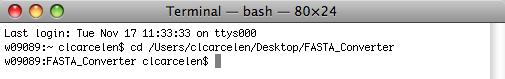
Hit Return.
Now, go back to the get_CDS_simpler.pl file you opened earlier in SubEthaEdit. Select " ./get_CDS_simpler.pl < input_filename > output_filename " as shown below and copy it into Terminal.
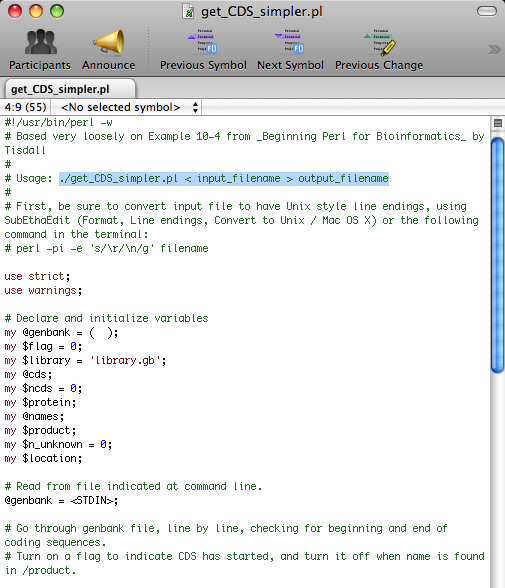
Replace " input_filename " with the the name of the file you wish to convert (this should be the name of the file you dragged into the FASTA_Converter folder earlier). The best way to do this is to open the folder and copy and p paste the file name into Terminal. Then, replace " output_filename " with a descriptive file name for the output file which will be in FASTA format.
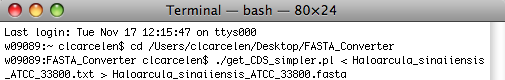
IMPORTANT NOTE: You must conserve the script exactly or errors will occur. Be sure to conserve the < > characters as well as any spaces in between them and the file names you enter. This is a simple mistake that can keep the entire program from running correctly.
Now, hit Return.
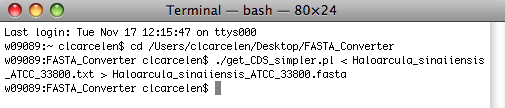
The Terminal window should resemble the one shown below, and a new file containing the file name you entered in Terminal should appear in the FASTA_Converter folder.
How to Download the Proteome Comparison Program
First, download the linked ZIP file. The icon pictured below should appear on your desktop, or in the folder you choose to download it to.
![]()
Click on the icon to unzip the contents. The folder icon pictured below should appear on your desktop once the file has been unzipped.
![]()
Once the folder is selected, the following files will appear in a window.
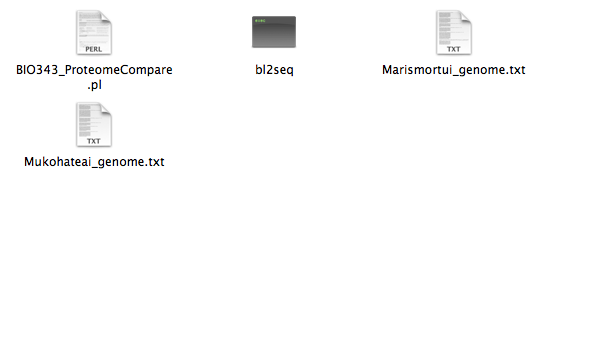
Select the ' BIO343_ProteomeCompare.pl ' file.
The file will open in SubEthaEdit, a real-time editing program designed for Mac OS X.
Follow the link below for a detailed tutorial on how to run the whole genome comparison using this program.
Pairwise Genomic Comparison Program Tutorial
This webpage was created by Claudia M. Carcelen for a Genomics class at Davidson College (2009).