This is a web page for an undergraduate course at Davidson College.
RHEUMATOID ARTHRITIS
Elizabeth Super
Dr. Malcolm Campbell
Biology 307: Immunology
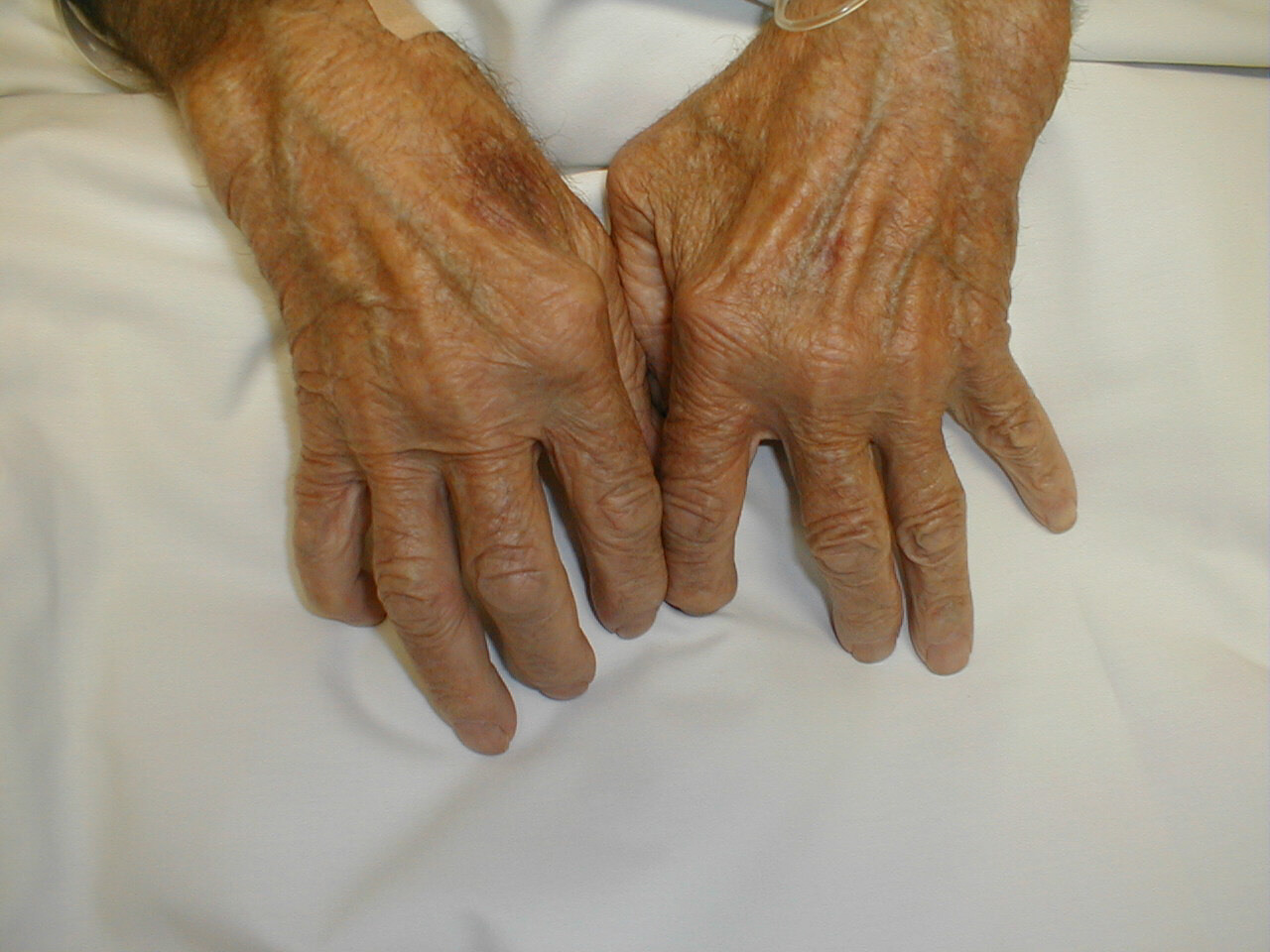
Fig. 1 Hands of a patient with Rheumatoid Arthritis. Rheumatoid arthritis is an inflammatory autoimmune disease that is characterized by the destruction of joint cartilage and inflammation of the synovium (joint fluid) (Abbas et al., 1994). The joints of the extremities, particularly the metacarpophalangeal, interphalangeal, and wrist joints, are the primary diseased sites (Bach, 1982).
Permission to use this image requested from charles.goldberg@med.va.gov <charles.goldberg@med.va.gov> on 23 April 2003. http://medicine.ucsd.edu/clinicalmed/upper.htm
Introduction to Autoimmune Diseases and Rheumatoid Arthritis (RA)
Autoimmune diseases occur when a self-antigen induces a specific adaptive immune
response. Although the adaptive immune response is normally able to clear foreign
antigens from the body, an adaptive response against a self-antigen cannot entirely
eliminate the antigen; therefore, a sustained response occurs, which can lead
to chronic inflammation and tissue damage. Autoimmune responses are initiated
in the same way as normal adaptive immune responses: with the activation of
antigen-specific T-cells. However, in autoimmunity, the T-cell response is directed
against a self-antigen. TH1 (T helper type 1) cells can cause tissue damage
by cytotoxic T-cell responses or by activating macrophages; TH2 cells can activate
self-reactive B cells to produce autoantibody responses (Janeway et al., 2001).
Rheumatoid arthritis (RA) is an inflammatory autoimmune disease that is characterized by the destruction of joint cartilage and inflammation of the synovium (joint fluid) (Abbas et al., 1994). The joints of the extremities, particularly the metacarpophalangeal, interphalangeal, and wrist joints, are the primary diseased sites, but as the disease progresses, the larger joints, especially the knees, can also be affected. The disease is an inflammatory arthritis that involves stiffness, pain, swelling, and erythema. As the disease progresses, osteoporosis of the contiguous bone, destruction of joint cartilage, bone resorption, and displacement by ankylosis occur. Subcutaneous nodules may also form at pressure points, particularly on the anterior side of the forearms, in 15-20% of reported cases. Systemic complications include damage to various organs (lungs, pleura, pericardium, myocardium, eyes, and central nervous system) due to inflammatory reactions. The outcome of the disease varies among patients, ranging from complete recovery to total incapacitation within years of the onset of the disease. In the most severe cases, vasculitis may occur. Women represent 75% of the reported cases of RA. There are two major peaks of incidence with regard to age: one between 30 and 40 years and one between 50 and 60 years (Bach, 1982).
Early experimental work focused on the importance of B-cells in the pathology of RA. Later interest has focused on T-cells. Observations that RA is strongly associated with certain MHC (major histocompatibility complex) alleles and that activated T-cells are found in affected joints have indicated that much focus should be given to cell-mediated immune responses. Whether activated T-cells initially respond to a microbial antigen, superantigen, or self-constituent is unclear to date (Chini et al., 2002).
Immunology and the Causes of RA
The development of lesions in rheumatoid arthritis appears to include both cell-mediated
and humoral responses. Early research aimed at identifying the cells present
in the inflamed synovium, and it has been concluded that CD4+ T-cells, activated
B-lymphocytes, and plasma cells, as well as well-formed lymphoid follicles with
germinal centers (in more severe cases), are present in the synovium of patients
diagnosed with RA (Abbas et al., 1994). T-cells are the dominant cell type in
the synovial filtrate of patients with RA, and there is at least a partial therapeutic
effect of T-cell depletion for these patients (Berner et al., 2000). Current
understanding of RA suggests that TH1 cells that are specific for a particular
antigen (which has yet to be identified) are present in the joints of people
with RA. This antigen activates T-cells to release lymphokines that cause local
inflammation at the joints. The clinical manifestations of this inflammation
include swelling, accumulation of polymorphonuclear leukocytes and macrophages
at the site of inflammation, cartilage damage, and, thus, destruction of the
joint (Janeway et al., 2001).
T-cells are the dominant type of cell that infiltrates the synovial membrane in RA. In many patients, the lymphocytes that infiltrate the tissues are organized into follicles that are structurally similar to germinal centers, which strongly supports the idea that RA is an antigen-driven immune response. Recent studies, however, have focused on the possibility that T-cells have an alternative role besides antigen recognition in the joints. Irregularities are found in the global and synovial T-cell receptor (TCR) repertoire of RA patients. The T-cell repertoire shows an emergence of clonal T-cell populations. CD4 T-cell clones are present uniformly in circulation and infiltrate into synovial lesions. CD8 T-cells clones are also found in patients with RA; although these clones are not limited to RA patients, they are larger and more frequent in people with the disease. CD4 T-cells are autoreactive to ubiquitous antigen, do not express the CD28 molecule, and do not require costimulatory signals to secrete cytokines. A study performed at the Mayo Clinic in 2002 found that RA is associated with a generalized defect in diversity maintenance of the TCR repertoire. This results in clonal expression of peripheral T-cells and repertoire contraction. Of significance, the researchers found that these irregularities involve memory and naïve T-cells, which suggests that the abnormality is a defect in T-cell homeostasis, rather than a consequence of antigen-recognition in the synovium. Specifically, the study showed that the CD4 TCR beta-chain diversity is limited in RA patients. Loss in diversity leads to the insufficient influx of ‘novel’ T-cells. Peripheral T-cells respond by replicating vigorously to compensate for the loss of new production, and clonal populations are formed. The multiclonal T-cells proliferation and the associated repertoire contraction have important implications for the course of the disease. It appears that the T-cell clones recognize a ubiquitously distributed autoantigen in RA patients. A recent study by Kouskoff et al. describes that transgenic mice expressing a TCR that recognizes self-MHC molecules develops arthritis that resembles RA but no other autoimmune diseases. To date, however, the autoantigen remains to be elucidated (Wagner et al., 1998).
Unusual distortions of the naïve T-cell repertoire have also been examined. These defects have led researchers to conclude that abnormal T-cell development and differentiation occurs in RA; autoimmunity could be caused by the inappropriate development and maturation of T-cells. RA patients appear to have fewer naïve T-cells with atypical phenotypes as compared to controls; these data suggest abnormal T-cell proliferation and phenotypic differentiation occur in response to inflammatory stimuli in patients with RA. These atypical cells appear to have a reduced threshold for activation and may bypass lymph nodes in favor of peripheral sites, which, during an infection, can lead to autoreactivity (Ponchel et al., 2002). Following this line of thought, hyperfunctioning T-cells could secrete local mediators in the synovial fluid, leading to arthritis. This hypothesis is supported by the fact that numerous T cells are found in the synovium of affected individuals. Also, injection of T-cell culture supernatants can induce inflammatory arthritis that resembles the clinical manifestations of RA. In this scenario, immune complex formation may be caused by T-cell hyperfunction through a helper effect, and the immune complexes would cause certain symptoms, including nodules and vasculitis (Bach, 1982).
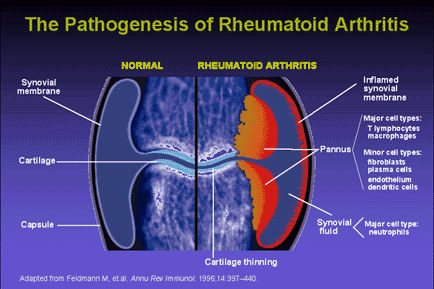
Fig. 2 The Pathogenesis of Rheumatoid Arthritis. The development of lesions in rheumatoid arthritis appears to include both cell-mediated and humoral responses. CD4+ T-cells, activated B-lymphocytes, and plasma cells, as well as well-formed lymphoid follicles with germinal centers (in more severe cases), are present in the synovium of patients diagnosed with RA. T-cells are the dominant cell type in the synovial filtrate of patients with RA. Cytokine secretion by activated T-cells leads to an inflamed synovium and the formation of pannus (Abbas et al., 1994).
Permission to use this image requested from kineret@amgen.com on 23 April 2003. http://www.kineretrx.com/professional/slide_center.jsp?slideID=2
Cytokines
Numerous cytokines have been detected in the synovial fluid of patients with
RA; these include interleukin-1 (IL-1), IL-8, tumor necrosis factor-alpha (TNF-alpha),
and interferon-gamma (IFN-gamma). These cytokines activate synovial cells to
produce hydrolytic enzymes, including collagenase, that cause destruction of
the cartilage, ligaments, and tendons on the joints. These cytokines are presumably
produced as a result of local activation of T-cells and macrophages. Researchers
have not yet identified a specific T-cell specificity or an initiating antigen
that leads to the clinical manifestation of RA (Abbas et al., 1994).
Two clinically important cytokines released in the synovium are IL-1 and TNF-alpha. Both cytokines increase the production of cyclo-oxygenase-2 (COX-2), nitric oxide, adhesion molecules, IL-6, chemokines, and collagenases. Both IL-1 and TNF-alpha stimulate the production of one other. IL-1 contributes to increased osteoclast activation and angiogenesis, and TNF-alpha increases apoptosis. The actions of these and other cytokines lead to the clinical manifestations of the disease. For example, IL-2 activates monocytes and macrophages, which leads to inflammation; it induces fibroblast proliferation, which causes synovial pannus formation; it activates chondrocytes, which leads to cartilage destruction; and it activates osteoclasts, which causes bone resorption (Kineret, 2001).
The balance between TH1 and TH2 cytokines is thought to have an important role in the outcome of autoimmune diseases. Recent studies focus on understanding the mechanisms involved in the development of TH1 proinflammatory effector cells that secrete IFN-gamma and the TH2 effector cells that secrete cytokines that downregulate the inflammatory process, such as IL-4. A study performed at the Turku University in Finland compared the production of IFN-gamma and IL-4 by CD4+ and CD8+ cells in both the synovial fluid (SF) and peripheral blood (PB) of patients with rheumatoid arthritis. The study concluded that the frequency of IFN-gamma-producing CD4+ and CD8+ cells was significantly increased in the SF as compared to the PB. The expression of IL-4 in both SF and PB T-cells was similar; however, the majority of IL-4-producing cells in SF were of the TH1 phenotype, while TH2 cells predominated in PB. These results suggest that patients with RA have an increased amount of inflammatory-inducing cytokines as well as a predominant number of TH1 cells in their synovial fluid. TH2 differentiation-inducing agents (such as IL-4) that are present do not seem to play a role in reversing the inflammatory process in the joint by shifting the balance of T helper cells in favor of TH2 cells. Possible future research involves injecting TH2 differentiation-inducing agents to shift the balance of T-helper cells to favor TH2 cells, which would produce anti-inflammatory cytokines and possibly reverse the disease symptoms (Isomaki et al., 1999).
Immune Complexes
Rheumatoid arthritis not only involves TH1 cells responding to self-antigens,
but it also involves antibodies (Janeway et al., 2001). RA leads to vasculitis,
which is most likely caused by the formation of immune complexes. Although the
nature of the antigen and the specific antibodies involved remain subjects for
debate, it is known that autoantibodies, called rheumatoid factors, are involved
(Abbas et al., 1994). Rheumatoid factors are anti-immunoglobulin antibodies.
In RA, the rheumatoid factors are usually IgM anti-IgG autoantibodies that react
with the Fc fragment of altered IgG (after combination with antigen or aggregation)
(Bach, 1982). The fact that rheumatoid factors have been isolated from the joints
of patients with RA provides evidence for a T-cell dependent, antigen-driven
B-cell response against the Fc portion of IgG, and the subsequent formation
of IgM:IgG immune complexes leads to tissue damage (Janeway et al., 2001). However,
the presence of rheumatoid factors is not specific for RA since they are also
present in 5% of normal subjects (Bach, 1982).
The existence of rheumatoid factors and immune complexes in association with RA suggests that a chronic antigenic stimulation is present in patients with RA. Many studies have attempted to identify a causative infectious agent in the synovium, but a single synovial joint antigen remains to be identified (Bach, 1982).
Susceptibility to RA
Susceptibility to RA is strongly linked to the human leukocyte antigen-DR4 (HLA-DR4)
haplotype and more weakly linked to the DR1 and DRW1D haplotypes. In each of
these alleles, the amino acid sequences from the sixty-fifth to the seventy-fifth
positions of the beta chain are nearly identical, and these residues are located
in the peptide-binding clefts of the HLA molecules; this suggests that they
affect either antigen presentation or T-cell recognition, although their role
in RA is not yet certain (Abbas et al., 1994).
Extensive studies have been done among different ethnic groups that have shown an association between the development of RA and the third hypervariable region of the major histocompatibility complex (MHC) HLA-DR beta 1 (DRB1) locus, which has been implicated in the protection to RA. A study performed by the Mayo Clinic found that the arthritogenic peptide-presenting molecule is HLA-DQ. The study concluded that the development of RA depends on three factors: expression of the susceptible DQ allele, expression of a nonprotective DRB1 allele, and environmental factors that trigger autoimmune processes (Zanelli et al., 1995).
There is much genetic heterogeneity in RA; most individuals who carry the known susceptibility alleles for RA do not get the disease, and, depending on the ethnicity of the population, many patients who manifest the disease do not carry the susceptibility alleles. Despite this variation, the link between human leukocyte antigen (HLA) DR-4 alleles and RA is one of the strongest examples of HLA risk genes among autoimmune disorders. The HLA-DR4 alleles DRB1*0401 and DRB1*0404 are the major susceptibility alleles associated with RA. These alleles are thought to have a role in the induction of autoreactivity by thymic selection or T-cell activation in the periphery. Two mechanisms have been proposed to explain this occurrence: interaction with the T-cell receptor (TCR) through the binding and presentation of unique peptides on HLA molecules or interaction of the MHC DRB1*04 molecule directly with the TCR (Chini et al., 2002).
MHC class II molecules are involved in the presentation of peptide to TCRs. Allelic variation among class II molecules allows for unique peptide-binding motifs, which accounts for the differences in peptides presented by a particular MHC allele. Binding motifs that are unique to disease-associated alleles may cause autoimmunity by influencing thymic selection and the T-cell repertoire. Likewise, they can allow the presentation of arthritogenic peptides in the periphery. The hypothesis that particular class II alleles can present unique peptides that lead to arthritis has been examined, but a unique antigen or peptide has not yet been clearly identified (Chini et al., 2002).
Intracellular antigen processing also influences the ability of a class II molecule. Alteration in the level of HLA DM in the thymus of RA patients could lead to the establishment of autoreactive T-cells during thymic selection. Decreased levels of DM expression in the periphery could cause the presentation of self-peptides that were not present during negative selection, which could cause autoimmunity. It has been found that levels of mRNA and DM protein expression are diminished in peripheral B-cells of RA patients compared with wild-type controls (Chini et al., 2002).
Also notable is the level of MHC:peptide complexes available to interact with a T-cell to cause T-cell activation. Studies have shown an upregulation of DR4 and DR1 in patients with RA. The surface expression of DR molecules on peripheral B-cells was examined, and an increase in DR expression was seen in RA patients compared to controls. Increased density of DR4 on the surface of an antigen-presenting cell (APC) can cause the activation of T-cells with low affinity peptides. With regard to RA, if a high level of DR4 is present on an APC, a low affinity self-peptide may lead to T-cell activation (Chini et al., 2002).
The HLA DR alleles associated with RA share a region within the third hypervariable region on the DR-beta chain. The sequence is called the shared epitope and is located on the alpha helix of the DR-beta chain, which allows it to interact with the TCRs. The contact between MHC and TCR may lead directly to autoreactivity. It has also been proposed that MHC molecules can actually be presented as self-peptides to APCs. These peptides would be available in the thymus leading to selection of T-cells susceptible to RA. They would also be available in the periphery to mimic a foreign peptide that has initiated an immune response, which would lead to persistent inflammation (Chini et al., 2002).
CD40 ligand on CD4+ T-cells as a Marker for Disease
CD40 ligand (CD40L) is a T-cell surface glycoprotein that is expressed transiently
on activated (but not resting) CD4+ T-cells. It binds to CD40, a tumor necrosis
factor receptor (TNFR) that is present on B cells, vascular endothelial cells,
monocytes and macrophages, and dendritic cells and fibroblasts. CD40L-CD40 interactions
are involved in humoral and cell-mediated immune responses. When activated CD40L+CD4+
T-cells are ligated to CD40 expressed on endothelial cells, upregulation of
certain adhesion molecules leads to increasing leukocyte migration and diapedesis.
CD40L-CD40 signaling on dendritic cells and macrophages induces the inflammatory
cytokines. Ligation of CD40L+CD4+ T-cells with CD40 on B-cells induces B-cell
proliferation and differentiation, isotype switching, and formation of memory
B cells (Berner et al., 2000).
The activation-induced T-cell antigen CD40L may be an important marker of disease
activity for patients with RA. The expression of CD40L by CD4+ T-cells is significantly
increased in RA patients as compared to wild-type controls. In RA, CD40-CD40L
interaction regulates the production of IL-12 on dendritic cells, which is required
for the initiation of TH1 type responses (RA has been identified as a TH1 mediated
disease). Also CD40L+ T-cells that infiltrate the joints of patients with RA
can interact with CD40+ synovial fibroblasts to cause their proliferation and
the upregulation of CD54, which helps to recruit more inflammatory cells and
increases the production of TNF-alpha (an inflammatory cytokine). Thus, the
increased expression of CD40L correlates with increased disease activity because
of prolonged activation of T-lymphocytes and the prolonged inflammation. The
finding that CD40L is hyperexpressed in RA patients has therapeutic significance.
Anti-CD40L antibodies may prove to be a significant immunotherapeutic approach
for patients with RA (Berner et al., 2000).
Treatments
Anti-TNF-alpha Treatments
Cytokines are protein mediators that are implicated in nearly all biological
processes, including cell growth, differentiation, inflammation, and immunity.
In patients with RA, nearly all cytokines are expressed in RA tissue in a continuous
fashion. Researchers previously thought that targeting the blockade of a particular
cytokine would be an ineffective therapeutic treatment since, they supposed,
other cytokines would take over the role of the blocked cytokine. Research with
RA has led them to a different hypothesis. IL-1 has been shown to induce the
destruction of cartilage and bone, and five signals present in the RA synovium
(IFN-gamma, GM-CSF, TNF-alpha, immune complexes, and IL-1 itself) help to regulate
IL-1 production. However, a study done at the Kennedy Institute of Rheumatology
found that blocking TNF-alpha abolished IL-1 bioactivity. Anti-TNF antibodies
were also found to downregulate GM-CSF, IL-6, and IL-8. Researchers determined
that pro-inflammatory cytokines are co-regulated, and the key inflammatory cytokines
are TNF-alpha and IL-2. This led the way for the development of anti-TNF-alpha
treatments; both soluble-receptor antagonists and cytokine antibodies have been
developed as anti-TNF-alpha therapies (Feldmann et al., 1999).
Medications Commonly Used to Treat Rheumatoid Arthritis (Rang et al., 1995)
Four types are medications are commonly used to treat rheumatoid arthritis.
These include aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs),
disease-modifying antirheumatic drugs (DMARDs), immuno-suppressants, and corticosteroids
(glucocorticoids).
NSAIDs have three major types of effect. They are anti-inflammatory agents;
they have an analgesic effect (reduction of pain); and they have an antipyretic
effect (lowering of raised temperature). Examples of these drugs include, but
are not limited to, plain aspirin, buffered aspirin, ibuprofen (Advil ®,
Motrin IB ®), ketoprofen (Orudis®), naproxen (Naprosyn®), celecoxib
(Celebrex®), and rofecoxiv (Vioxx®).
Anti-inflammatory effects: The primary action of the drugs is to inhibit
arachidonate cyclo-oxygenase and, thus, to inhibit the production of prostaglandins
and thromboxanes. One type of cyclo-oxygenase, COX-2, is induced in activated
inflammatory cells and is the enzyme that produces the prostanoid inflammatory
mediators. NSAIDs reduce the components of inflammation that are caused by COX-2
action, which include vasodilation, edema, and pain. These drugs have no effect
on the processes that contribute to tissue destruction in RA; they simply reduce
the generation of toxic O2 products and inhibit lymphocyte activation.
Antipyretic effects: NSAIDs inhibit prostaglandin production in the
hypothalamus. During an inflammatory reaction, macrophages release IL-1, which
stimulates the generation of E-type prostaglandins in the hypothalamus, which
elevate the body temperature of the individual. NSAIDs, thus, act to reduce
body temperature when it is raised above normal levels.
Analgesic effects: NSAIDs are effective against pain that is caused
by prostaglandins acting on nociceptors (ie. pain associated with inflammation
or tissue damage). Decreased prostaglandin production leads to less sensitization
of nociceptic nerve endings to the inflammatory mediators, bradykinin and 5-hydroxytryptamine
(Rang et al., 1995).
DMARDs are distinct from NSAIDs in that they do more than simply alleviate the
symptoms of RA. They are used to alter the course of the disease and prevent
joint and cartilage destruction. Three types of DMARDs are widely used; these
include gold compounds (Myochrysine®, Ridaura®), penicillamine (Cuprimine®,
Depen®), and chloroquine (Plaquenil®). Although their mechanisms of
action have not been fully elucidated, their effects have had a profound effect
on RA patients.
Gold Compounds: Gold compounds help to stop the progression of bone
and joint damage in RA. Pain and joint swelling are reduced when these drugs
help to reduce the concentration of rheumatoid factor. Although the exact mechanism
of action is not fully understood, any or all of the following effects can contribute
to the mechanism: inhibition of mitogen-induced lymphocyte proliferation, reduction
of lysosomal enzymes, reduction in the production of toxic O2 metabolites from
phagocytes, inhibition of chemotaxis of neutrophils, and reduction in IL-2 production.
Penicillamine: Penicillamine prevents the maturation of newly synthesized
collagen. Although the mechanism of action is not fully understood, it has been
observed that joint swelling subsides, nodules disappear, and IL-1 production
is reduced. About 75% of patients with RA respond to this treatment.
Chloroquine: Anti-malarial drugs cause remission of RA but do not stop
bone destruction. They inhibit mitogen-induced lymphocyte proliferation and
decrease leukocyte chemotaxis. They also reduce IL-1 production (Rang et al.,
1995)
Immuno-suppressants, which are considered DMARDs, restrain an overly-active
immune system. These drugs include cyclosporine (Sandimmune®, Neoral®)
and cytotoxic agents such as azathioprene (Imuran®).
Cyclosporine: Cyclosporine suppresses both cell-mediated and humoral
responses. It acts on T-lymphocytes at the induction stage to stop clonal proliferation.
The transduction pathway for lymphokine synthesis is inhibited, mainly the production
of IL-2. It also inhibits IL-2 receptor expression on T-cells that respond to
IL-2. The induction of cytotoxic T-cells is also inhibited. B-cell responses
are suppressed because lymphokine synthesis and secretion from activated T-cells
is inhibited.
Azathioprine: Azathioprine is metabolized to give mercaptiopurine,
which is a purine analogue that inhibits DNA synthesis. This drug depresses
both cell-mediated and humoral responses since it inhibits clonal proliferation
by a cytotoxic action on dividing cells (Rang et al., 1995).
Corticosteroids have anti-inflammatory and immunosuppressive effects. They include prednisone (Deltasone®, Orasone®), hydrocortisone, dexamethasone, and methylprednisolone (Medrol®). These drugs reduce vasodilation and decrease fluid exudation. In areas of acute inflammation, they decrease the number and activity of leukocytes (decreased action of T-helper cells and reduced clonal proliferation of T-cells due to decreased production of IL-2; decreased release of monocytes and increased number of neutrophils from the bone marrow). In areas of chronic inflammation, the activity of mononuclear cells is decreased. In lymphoid areas, there is decreased clonal expansion of T- and B-cells and decreased action of cytokine-secreting T-cells. These drugs decrease the production of many cytokines, including IL-1, IL-2, IL-3, IL-4, IL-5, Il-6, IL-8, TNF-alpha, and GM-CSF. They also help to decrease the complement component in the blood. Overall, they reduce chronic inflammation and autoimmune reactions. Glucocorticoids act by interacting with intracellular receptors; the steroid-receptor complex interacts with DNA to modify gene transcription. These drugs perform their anti-inflammatory and immunosuppressive actions as follows: inhibition of transcription of the gene for COX-2; blockage of vitamin-D3-mediated induction of the osteocalcin gene in osteoclasts and modification of the transcription of the collagenase gene; and increased synthesis of an anti-inflammatory mediator protein (lipocortin 1) which inhibits phospholipase A2 and blocks the production of platelet-activating factor (Rang et al., 1995).
Works Cited
Abbas, A., Lichtman, A., Pober, J. 1994. Cellular and Molecular Immunology.
Philadelphia, PA: W. B. Saunders Company. p. 403-404.
Bach, Jean-Francois. 1982. Immunology. New York, NY: John Wiley & Sons,
Inc. p.
814-819.
Berner, B., Wolf, G., Hummel, K., Muller, G., Reuss-Borst, M. 2000. Increased
expression of CD40 ligand on CD4+ T cells as a marker of disease activity in
rheumatoid arthritis. Annals of the Rheumatic Diseases. 59: 190-195.
Chini, L., Bardare, M., Cancrini, C., Angelini, F., Mancia, L., Cortis, E.,
Finocchi, A.,
Riccardi, C., Rossi, P. 2002. Evidence of clonotypic pattern of T-cell repertoire
in synovial fluid of children with juvenile rheumatoid arthritis at the onset
of disease. Scandinavian Journal of Immunology. 56 (5): 512-517.
Feldmann, M., Bondeson, J., Brennon, F., Foxwell, B., Maini, R. 1999. The rational
for
the current boom in anti-TNF-alpha treatment. Is there an effective means to
define therapeutic targets for drugs that provide all the benefits of anti-TNF-alpha
and minimize hazards? Annals of the Rheumatic Diseases. 58: 127-131.
Isomaki, P., Luukkainen, R., Lassila, O., Toivanen, P., Punnonen, J. 1999.
Synovial
fluid T cells from patients with rheumatoid arthritis are refractory to the
T helper type 2 differentiation-inducing effects of interleukin-4. Immunology.
96 (3): 358-364.
Janeway, C., Travers, P., Walport, M., Shlomachik, M. 2001. Immunobiology.
New
York, NY: Garland Publishing. P. 501-502, 520-521.
“Kineret.” Amgen, Inc. (2001): n. pag. Online. Internet. 15 April
2003. Available
http://www.kineretrx.com/professional/slide_center.jsp?slideID=2.
Ponchel, F. et al. 2002. Dysregulated lymphocyte proliferation and differentiation
in
patients with rheumatoid arthritis. Blood. 100 (13): 4550-4556.
Rang, H., Dale, M., Ritter, J., Gardner, P. 1995. Pharmacology. New York, NY:
Churchill Livingstone. p. 246-264, 261-264, 435-445.
Wagner, U., Koetz, K., Weyland, C., Goronzy, J. 1998. Perturbation of the T
cell
repertoire in rheumatoid arthritis. Proc. Natl. Acad. Sci. 95 (24): 14447-14452.
Zanelli, E., Gonzalez-Gay, MA., David, CS. 1995. Could HLA-DRB1 be the protective
locus in rheumatoid arthritis? Immunology Today. 16 (6): 274-278.
E-mail questions or comments