This web page was produced as part of an undergraduate assignment at Davidson College.
Sjogren's Syndrome
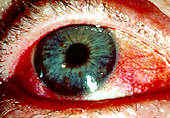
Figure 1. This red, dry eye is one common symptom of Sjogren's syndrome.
Permission for use granted by Rolf Manthorpe, Head of the Sjogren's Syndrome Research Centre
Introduction to Sjogren's. Sjogren's syndrome (SS) is an autoimmune disease in which the body's immune system mistakes its moisture-producing glands for foreign invaders (Sjogren's Syndrome Foundation [SSF]:1999). It is characterized by "mononuclear infiltrates in the exocrine glands and numerous complications involving the peripheral and central nervous systems" (Mochizuki, et. al. 2000:1391). There are four main symptoms of the syndrome: fatigue, dry eyes, dry mouth, joint pain (Sjogren's Syndrome Research Center [SSRC], na). Two types of SS are typically diagnosed. Primary SS involves parotid and other salivary glands and the lacrimal glands in the eyes. This type is present without any other autoimmune disorder of the connective tissue. Secondary SS occurs in conjunction with an autoimmune, connective tissue disorder, and it includes all the symptoms of primary SS (SSF, 1999). There is no known cure.
History of SS. Henrik Sjogren was born in Sweden in 1899 and graduated from the Karolinska Institute of medicine in 1927. In 1930, he described five cases of keratoconjunctivitis sicca [KCS] and published a thesis on his findings with nineteen patients in 1933 (SSRC, na). Thirteen of these patients expressed both dry eyes and arthritis (Talal 1992:508). During World War II, the term "Sjogren's syndrome" was coined. In 1951, Sjogren published eighty cases of the syndrome, the second largest population of patients at the time (SSRC, na). According to Norman Talal, MD, the history of the syndrome can be divided into three phases: Clinical (1888-1950), immunologic (1950-1980), and molecular (1980-present). Sjogren's two important contributions to the understanding of this syndrome were the staining of corneal lesions with Rose Bengal and the introduction of the term "keratoconjunctivitis sicca." In 1954, Morgan and Castle determined that SS and Mikulicz disease (manifests as painless swelling of lacrimal, parotid, and submandibular glands; described in 1888) were one and the same. Since 1950, the syndrome has been studied immunologically, centering on humoral immunity, the rheumatoid factor, the relationship between SS and anti-Ro/SS-A and anti-La/SS-B, and the antinuclear factors, and more. Today, scientists are examining the possibility of a viral cause (Talal 1992: 508-9).
Clinical Aspects of SS. The main difference between primary and secondary SS (see Table 1) is the presence of a connective tissue disease in secondary SS. There is also a true overlap between SS and systemic lupus erythematosus [SLE]. The progression of secondary SS is also slower than in primary (Talal 1992:510).
Table 1. Distinguishing characteristics of primary and secondary Sjogren's syndrome (Talal 1992:510)
| Primary Disease
KCS and positive lip biopsy without another underlying rheumatic disease HLA-B8-DR3-positive Presence of antinuclear antibodies to Ro/SSA and La/SSB
|
| Secondary Disease
Evidence of accompanying rheumatoid arthritis (RA) or other connective tissue disease Immunogenetic and serologic findings of the accompanying disease (e.g. HLA-DR4-positive if RA)
|
There are approximately one to two million people living in the United States with SS, and most remain undiagnosed. The syndrome may present malignantly (lymphoma) or as a benign autoimmune disease. Clinically, SS presents variably (see Table 2).
Table 2. Clinical presentation of Sjogren's syndrome (Talal 1992:510)
| 1. Sicca complex -- dry eyes and dry mouth
2. Rheumatoid arthritis or other connective tissue disease 3. Salivary gland enlargement 4. Purpura -- nonthrombocytopenic; hyperglobulinemic, vasculitic 5. Renal tubular acidosis or other tubular disorder 6. Polymyopathy 7. Central or peripheral nervous system 8. Chronic liver disease 9. Chronic pulmonary disease 10. Lymphoma -- local or generalized 11. Immunoglobulin disorder -- cryoglobulinemia, macroglobulinemia |
The mean age is 50 and the majority (90%) of patients are women. A study by Anaya, et. al. concluded that clinical and serological indications are similar between men and women, with men showing more systemic manifestations and autoantibodies. This study also suggested that sex hormones may play a role in the severity of the disease, but this is still uncertain (Anaya, et. al.1995:748). A later study by Drosos, et. al. of Greek male and pediatric SS patients revealed, in contrast, that SS is rare among these populations. The study also showed that males tend to have less systemic manifestations and a lower frequency of autoantibodies. The resarchers hypothesized the difference between the studies may be due to genetic makeup of patients (Drosos, et. al. 199:334).
Parotid gland enlargement is present in about 50% of cases, and the mouth dryness is problematic for most patients (see Figure 2). Patients typically have difficulty chewing and swallowing and may not be able to wear dentures. Decreased tearing, redness, itchiness, and a
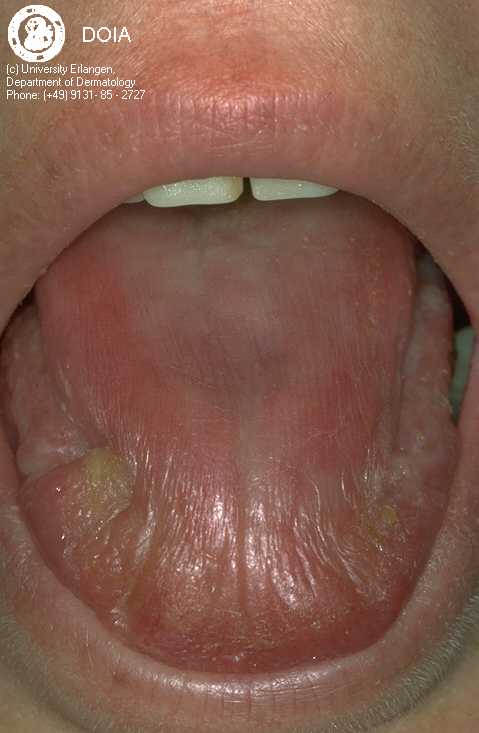
Figure 2. This dry tongue is from a 39 year old patient. Permission has been requested from Dermis for use of this picture.
"filmy" sensation are all common ocular complaints (see Figure 1). Dryness may also be present in the nose, pharynx, vagina, or skin. The syndrome may result in the development of glomerulonephritis, polymyositis, and central nervous system disease [CNS], although these are debated (Talal 1992:511). Recent studies indicate SS related CNS is due to cell-mediated neuronal injury resulting in inflammation (Lafitte 2000:413). Cirrhosis and hepatitis may also play a role is SS (Talal 1992:511).
The Immune System and SS. The primary autoantibodies in SS are anti-Ro/SS-A and anti-La/SS-B (See Table 3). About 70% of patients with SS are positive for anti-Ro/SS-A (35% of those with SLE). Autoantibodies for anti-La/SS-B are present in over 50% of SS patients and about 10% of SLE patients. This antigen is an RNA-binding protein and is associated with several viral transcripts including Epstein-Barr virus EBER RNA's (Chan and Andrade 1992, 552, 559). MA-I and p80-coilin have not been studied closely yet. According to Henriksson et al., an intron-located promoter switch leads to mRNA processing modification and gives the altered expression of the La/SS-B gene. This may be one way MHC's can express altered self-antigen and activate autoimmune T cells (Henriksson et al. 2000:146).
Table 3. Nuclear antigens and prevalence of autoantibodies in SS (Chan and Andrade 1992, 552)
| Desigation of Autoantigen | Prevalence of Autoantibody | Molecular Characteristics |
| anti-Ro/SS-A | 70% | Proteins, 60 and 52-kD, complexed with hY1-5 RNAs |
| anti-La/SS-B | 50% | Phosphoprotein, 47-kD, complexed with nascent RNA polymerase III transcripts |
| p80-coilin | 4% | Phosphoprotein, 80-kD, primarily restricted to the nuclear coiled body |
| MA-I | 8% | Nuclear proteins, approx. 200-kD, localized to the mitotic apparatus in dividing cells |
The infiltration of primarily CD4+ T cells into the lacrimal and salivary glands is the main cause of mouth and eye dryness among SS patients. Comparisons between peripheral blood lymphocytes [PBL] and salivary gland [SG] lymphocytes revealed distinct differences in phenotype and function. The SS SG lymphocytes produced such cytokines as IL-2, IL-10, and interferon-gamma, and DNA often shows oligoclonal rearrangement of the immunoglobulin [Ig] genes. The PBL cells did not have these features. SG epithelial cells are most likely participants in the immune response since the cells express class II HLA molecules and may secrete IL-1 and IL-6 (Fox and Kang 1992:517). The CD4+ lymphocytes are depleted in patients with SS. A study by Henriksson et al. showed, using ELISA, that anti-CD4 IgG antibodies are present in patients with primary SS, but no correlation could be made between anti-CD4 antibodies and CD4+ T lymphocytopenia. One potential cause of the autoimmune response is the presence of T cells with cryptic epitopes. These result from the inadequate presentation of certain self proteins on MHC molecules. This may then lead to autoreactive T cells (Henriksson et al. 2000:142, 145-6).
A study by Halse et al. determined the location of antibodies in the plasma and saliva of seventeen SS patients. The results showed increased levels of IgG, IgM, and IgA in the saliva, with the majority of patients having increased levels of IgG in both saliva and plasma. The increased levels of Ig in saliva suggests that antibodies are produced locally, but the levels may be due to extravasation due to inflammation in the area. It is known that at least part of the number of antibodies present are produced locally since there is a greater number of IgG's and IgM's in the salivary tissue of SS patients than in normal patients. The highest levels of all three isotypes were found in the plasma, suggesting that the majority of antibody production occurs in extra-glandular sites. The researchers presented the levels of "anti-Ro/SS-A and anti-La/SS-B antibodies as antigen specific antibody" in relation to the total level of each isotype. They found that 1/3 of the patients had levels of IgA and IgM in saliva more than twice greater than in plasma. This may be due to local production of the antibodies in the saliva. The study also showed that more IgA antigen specific antibodies were produced in the saliva than IgG. The presence of slightly greater numbers of IgA and IgM in saliva may be due to their ability to cross mucous membranes (Halse et al. 2000, 17). Excessive IgA has also been suggested as a causative agent for SS. Basset et al. determined that sialyl transferase [ST]6 and ST3 of the B cell are hyperactive and cause hypersialylation of IgA. Since and increased serum IgA, IgA-immune complexes, and rheumatoid factor are all present in primary SS, it is possible to suggest that these are due to B cell activation. The accumulation of serum IgA may lead to a pathogenic role for IgA in SS. It is not known whether the secretory form of IgA is also hypersialylated (Basset et al. 2000:310-11).
Further studies have revealed a key role of apoptosis in the destruction of glandular tissue. Fujihara et al. demonstrated that CD8+ T cells are located around the acinar epithelial cells. Most of these contain CD103, a unique integrin which apparently is involved in the apoptotic pathway of the epithelial cells. In lacrimal glands, the Fas/Fas ligand and perforin/granzyme B pathways were used for cell death. Fujihara et al. concluded that CD130, "by interacting with E-cadherin, mediates the adhesion between CD8+ T lymphcytes and acinar epithelial cells is SS and participates in inducing epithelial cell apoptosis, leading to a secretory dysfunction of exocrine glands" (Fujihara et al. 1999:2226). Tsunoda et al. determined that there is a diminished expression of CD59 on CD8+ T cells in both SS and SLE. These T cells were shown to be more susceptible to in vitro apoptosis than non-diminished CD59 T cells. This suggests that decreased expression of CD59 antigen may play a part in the apoptotic pathway seen in SS and SLE (Tsunoda et al. 2000:293). Patel and McHugh state that there is a larger amount of apoptosis seen in acinar and ductal epithelial cells than in the infiltrating lymphocytes. They also suggest that the initial apoptotic event (prior to lymphocyte infiltration) may have a viral source. In this case, the cells would stage a normal response to viral infection but would then be unable to control apoptosis, resulting in tissue damage and the clinical presentations of SS (Patel and McHugh 2000:119-20).
The presence of antineutrophil cytoplasmic antibodies [ANCA] in SS was studied by Font et al. in 1998 and is thought to be important in the manifestation of one complicationo of SS. Out of 82 patients expressing primary SS, 11% were positive for ANCA. Fifteen to thirty percent of the 82 patients also had vascular involvement (Raynaud's phenomenon), peripheral neuropathy, and cutaneous vasculitis, and a higher level of these manifestations were seen in the patients with ANCA. A complication of SS is inflammatory vascular disease [IVD] which has two subsets: MIVD and NIVD. Eighteen patients had MIVD which was found to be related to ANCA. Since it is possible that ANCA can cause vasculitic inflammatory lesions, it has been suggested that ANCA might be the causative agent in MIVD, but more studies must be done (Font et al. 1998:1290).
Treatment of SS. KCS, or dry eyes, is usually treated with artificial tears which must be used regularly and should be prescribed based on local environmental conditions. KCS can vary in SS patients. Some patients have only mild irritation while others may go blind. Punctual occlusion can be used in severe cases when the tear treatment has ceased being effective. This method involves occluding the punctual openings within the eye so as to prevent the artificial tears from draining as quickly. Surgical intervention may eventually be required. Surgical measures include the sewing of the lateral eyelids to prevent moisture evaporation (Fox 1992: 700-1).
Certain special toothpastes and oral gels can be prescribed to help treat dryness in the mouth. The toothpastes often use a galactose or sodium bicarbonate substrate to produce low levels of peroxide which can assist in the antibacterial action of the immune system. Numerous infections can also occur in a dry mouth, and these are treated by various methods suiting each case. Nasal dryness must be treated to prevent breathing through the mouth and further aggravation of dryness. Saline nasal sprays and lavage are used to keep the nasal passages moist and free from build-up (Fox 1992:702-3).
Dry skin and vaginal dryness are treated with moisturizers and lubricants. Estrogen replacement therapy in menopausal patients has not been shown to result in worsening of SS. Fatigue and depression are also common symptoms. Good sleep habits are generally initially prescribed for the former, while antidepressants lacking anticholinergic agents can be used for both (Fox 1992:706-7).
With regard to myalgias and arthralgias, anti-inflammatory medications are often employed. In certain patients, hydroxychloroquine has been used and was shown to decrease myalgias, arthralgias, and gland/node swelling. This treatment was probably successful due to the active inflammatory immune response which is a cofactor to myalgias and arthralgias. Systemic steroids are usually given to treat infiltration of other organs (hemolytic anemia, vasculitis, etc) (Fox 1992:707-8).
There is no cure for SS, but it is possible to treat and control symptoms of the syndrome. Treatments are also necessary to stop or slow the advancement of SS. Further studies are needed before it will be possible to consider an immune-related treatment.
NOTE. Patients with SS, primary or secondary, often have other family members with the syndrome. Anti-Ro and anti-La autoantibodies are associated with the HLA class II alleles and this fact may elucidate a possible genetic role in the development of SS. At this time, little is known about whether there is a definitive genetic cause for SS, but research is continuing in this area (Reveille and Arnett 1992:548).
SOURCES.
Anaya, Juan-Manuel and others. 1995. Primary Sjogren's syndrome in men. Annals of the Rheumatic Diseases 54: 748-51.
Basset, C and others. 2000. Enhanced sialyltransferase activity in B lymphocytes from patients with primary Sjogren's syndrome. Scandinavian Journal of Immunology 51: 307-11.
Chan, Edward K, and Luis E. C. Andrade. 1992. Antinuclear antibodies in Sjogren's syndrome. Rheumatic Disease Clinics of North America 18(3): 551-70.
Dermis Dermatology Internet Service. 1998. Sjogren's syndrome. Unversity of Erlangen Nurnberg. 6 April 200 <http://www.dermis.net/bilddb/diagnose/englisch/i710200.htm>
Drosos, Alexandros A. and others. 1997. Subgroups of primary Sjogren's syndrome: Sjogren's syndrome in male and paediatric Greek patients. Annals of the Rheumatic Diseases 56: 333-35.
Font, J. and others. 1998. Antineutrophil cytoplasmic antibodies in primary Sjogren's syndrome: Prevalence and clinical significance. British Journal of Rheumatology 37: 1287-91.
Fox, Robert I. 1992. Treatment of the patient with Sjogren's syndrome. Rheumatic Disease Clinic of North America 18(3): 699-709.
Fox, Robert I. and Ho-Il Kang. 1992. Pathogenesis of Sjogren's syndrome. Rheumatic Disease Clinic of North America 18(3): 517-38.
Fujihara, Tsutomu and others. 1999. Preferential localization of CD8+ (CD103)+ T cells around acinar epithelial cells with apoptosis in patients with Sjogren's syndrome. Journal of Immunology 163: 2226-35.
Halse, A.-K., M. C. Marthinussen, M. Wahren-Herlenius, and R. Jonsson. 2000. Isotype distribution of anti-Ro/SS-A and anti-La/SS-B antibodies in plasma and saliva of patients with Sjogren's syndrome. Scandinavian Journal of Rheumatology 29: 13-19.
Henriksson, G., R. Manthorpe, and A. Bredberg. 2000. Antibodies to CD4 in primary Sjogren's syndrome. Rheumatology 39: 142-47.
Lafitte, Catherine. 2000. Neurological manifestations in Sjogren syndrome. Archives of Neurology 57: 411-13.
Mochizuki, Akihide, Akito Hayashi, Shin Hisahara, and Shin'ichi Shoji. 2000. Steroid-responsive Deric's variant in Sjogren's syndrome. Neurology 2: 1391-2.
Patel, Y. I. and N. J. McHugh. 2000. Apoptosis: New clues to the pathogenesis of Sjogren's syndrome? Rheumatology 39(2): 119-21.
Reveille, John D. and Frank C. Arnett. 1992. The Immunogenetics of Sjogren's syndrome. Rheumatic Disease Clinic of North America 18(3): 539-50.
Sjogren's Syndrome Foundation. 1999. What is Sjogren's syndrome? 6 April 2000 <http://www.sjogrens.org/whatis.htm>
Sjogren's Syndrome Research Centre. Symptoms, diagnosis, and treatment. 6 April 2000 <http://www.medforsk.mas.lu.se/ssrc/symptoms.html>
Talal, Norman. 1992. Sjogren's syndrome: Hisorical overview and clinical spectrum of disease. Rheumatic Disease Clinic of North America 18(3): 507-15.
Tsunoda, S. and others. 2000. Diminished expression of CD59 on activated CD8+ T cells undergoing apoptosis in systemic lupus erythematosus and Sjogren's syndrome. Scandinavian Journal of Immunology 51: 293-99.
Go back to Heather's Immunology Home Page
![]()
Learn more about Immunology from the Davidson College Immunology Home Page
©Copyright 2000 Department of Biology, Davidson College, Davidson, NC 28036
Send comments, questions, and suggestions to: hebaker@davidson.edu