MacDNAsis Page
After using GenBank to locate the mRNA sequence for the human ubiquitin protein (chick here to see the original GenBank file), I used MacDNAsis program to analyze the sequence.
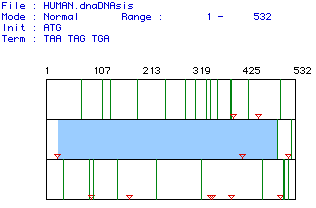
Fig. 1 The results of a MacDNAsis open reading frame analysis. All start and stop codons are represented by red triangles. Of the 532 base pairs and the three reading frames (one displayed per row) scanned, the largest open reading frame was found in the second row beginning at the 26 base pair and running to the 496th base pair, as indicated by the blue box. The original mRNA sequence can be found here.
MacDNAsis was also used to determine the molecular weight of the protein that would be transcribed by the open reading frame sequence in the mRNA. The program predicts that the transcribed protein would weigh 17964.08 Daltons.
In order to determine, more about this ubiquitin protein, specifically whether the protein has a transmembrane domain, a Kyte-Doolittle analysis was conducted. This program measures and scores the hydrophobicity of window of amino acids. This is done for the entire amino acid sequence. Windows with a positive score are hydrophobic, while negative windows are hydrophilic. If an window or set of windows have a score higher than 1.8, they are predicted to be part of a transmembrane domain.
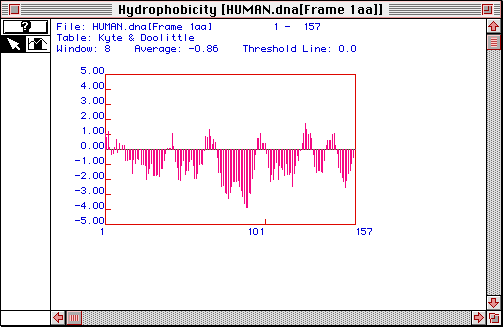
Fig. 2 The results of a Kyte-Doolittle analysis of the human ubiquitin protein. A window size of 8 amino acids was used. Most of the windows do not approach the 1.8 cut-off value. However, there is one part of the sequence that might be part of a transmembrane domain. Around amino acid # 125, there is a peak that might be greater than 1.8. Therefore, the results of the Kyte-Doolittle analysis predicts a possible transmembrane domain.
Another analysis called the Hopp and Woods was used to determine if there are any possible antigenic regions of the protein. This test is very similar to the Kyte-Doolittle except that it is the degree of hydophilicity in a window that is used to predict antigenic regions. The theory behind this test is that antigenic regions, regions that are not within or surrounded by a membrane, should be hydrophilic.
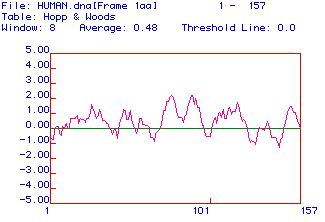
Fig.3 The results of a Hopp and Woods analysis. A window width of 8 amino acids was used. The more positive the peaks are the greater the hydrophilicity of that region of the protein. Most of the protein is predicted to be antigenic because the threshold line is 0.0. Therefore, based on this and the previous analysis, I would predict that most of the protein (amino acids 1 to approximately 125) is in the cytocil, around 125 the protein has transmembrane domain but the last part of the protein reemerges on the cytocilic side.
The final analysis performed to learn more about the human ubiquitin protein was a Chou, Fasman and Rose prediction of the proteins secondary structure.
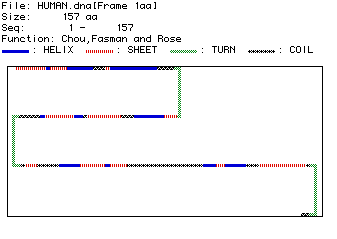
Fig. 4 The results of the Chou, Fasman and Rose secondary structure prediction. The structure of the amino acid sequence is indicated by the color coded legend. This protein appears to have three large turns that separate regions of interspersed helix, sheet and coil formations. This secondary structure does not agree exactly with the hypothesized transmembrane domain and two cytocilic regions, but it does not exclude the possibility.
Once I had completely analyzed the human ubiquitin, it was time to compare this sequence to the ubiquitin amino acid sequences of four other species Chlamydomonas, C. elegans, Drosophila and Mus musculus. The Higgins program generated the best fit so it was used.
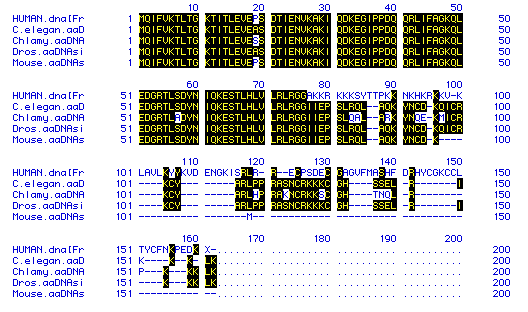
Fig. 5 The results of the Higgins comparison of amino acid similarity among four species. The species for each row is indicted on the left and matches are highlighted in black. Hyphens were added to increase the matching. The comparison indicates that the amino acid sequence is relatively conserved for the first part of the protein. However, in later portions, the species diverge. The human sequence appears to be the most different.
Finally, the Higgins analysis was used to construct a phylogenetic tree of the five species, Chlamydomonas, C. elegans, Drosophila, Mus musculus and Homo sapiens based on their ubiquitin amino acids sequence similarities.
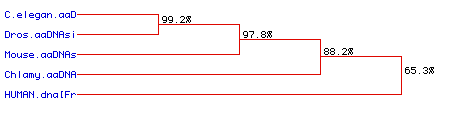
Fig. 6 The results of the Higgins phylogenetic predictions. The amino acid sequences of C. elegans and Drosophila ubiquitin protein are highly conserved. The mouse, Mus musculus, was also similar, but slightly less so. The Chlamydomonas ubiquitin diverged further, but the most distinct amino acid sequences was that of human ubiquitin.