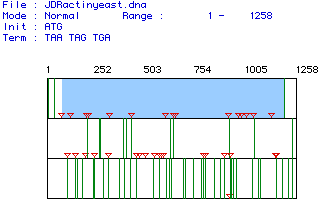
Figure 1. (Above) The open reading frame of yeast actin spans nucleotides 73-1200. The open reading frame is highlighted in blue, down arrows indicate start codons (ATG), and green lines indicate stop codons. The open reading frame is predicted by the longest uninterrupted stretch between start and stop codon.
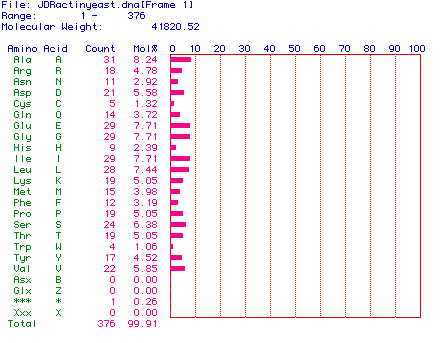
Figure 2. (Above) The molecular weight of yeast actin was determined to be 41820.52 daltons. Amino acids are listed in green with their corresponding one letter codon, their respectives abundances in actin are listed in red with each amino acid's percentage of the molecular weight. Molecular weight was determined by actin sequence analyzation.
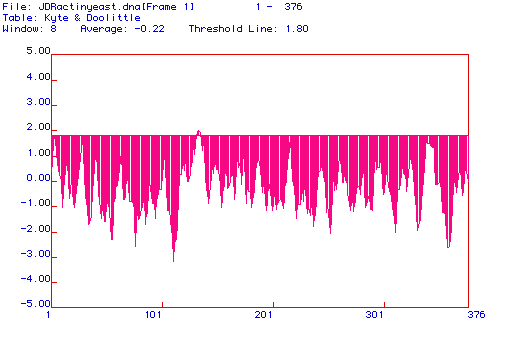
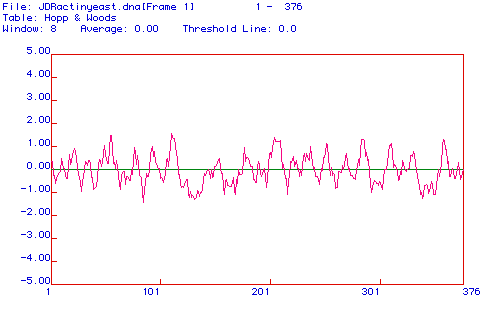
Figure 4. (Above) Hopp & Woods hydrophilicity, or antigenicity, plot of yeast actin cDNA. The Hopp and Woods computer prediction indicates the most probable epitope for antibody binding, based on hydrophilicity predictions from nucleotide analyzation. The greatest positive peaks indicate the most hydrophilic regions, thus the most likely regions to be extracellular and exposed to antibodies. Future research should concentrate on residues 102-120 and 195-210 for epitope sites, as they are the among the strongest peaks. Epitope searches should not be limited to these residues, however, as other peaks are relatively strong as well.

Figure 5. (Above) The predicted secondary structure of 376 amino acids within yeast actin cDNA. H represents alpha helices, S indicates Beta pleated sheets, t represent turns, and c indicates coils in the secondary structure of yeast actin. This analysis shows the repeating nature of helices and sheets for multiple amino acids. Turns and coils, while repeating at times, also have solitary occurances for a single amino acid. The secondary (as well as primary, tertiary, and quaternary) structure, or shape, of a protein is essential to its function. Secondary structure is determined by molecular interactions and binding patterns within the molecule.

Figure 6. (Above) Actin amino acid comparison between (from top to bottom) mouse (Mus), yeast (Schizosaccharomyces), sea hare (Aplysia), fruit fly (Drosophila), and nematode (C. elegans). Yellow letters on black indicate retained amino acids between the organisms and blue letters on white indicate unretained amino acids. Dashes indicate spaces that were added to maximize homology between the organisms. The high amount of yellow characters on black throughout this sequences represents a great deal of homology between there five organisms.
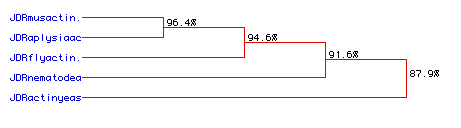
Figure 7. (Above) A predicted phylogeny, based on the previous amino acid comparison (Figure 6) of the five survey organisms. Red lines represent predicted relationships between organsisms based on examination of the actin protein. Percentages quantitate the amount to which each organism is related to the next "closest" organism. 24% is considered the minimum point at which two organism are homologous; homolgy is considered random for any values under 24%. Organisms are listed from bottom to top in order of relative increasing "complexity."
View my Genbank Search of Actin
Return to Jeremy's Home Page.
Now you can return to the Molecular Biology Home Page.
or to the Davidson College Home Page.