MacDNAsis: analysis of DNA binding protein
With the MacDNAsis program, a series of analyses were conducted on the cDNA sequence of the human DNA binding protein, which was obtained from the Genbank at the National Center for Biotechnology Information. To begin the analyses, the open reading frames (ORFs) of the cDNA were examined to determine the largest ORF in the sequence. The neucleotide sequence of the largest ORF was then translated, and the amino acid content was used to predict the molecular weight of the protein. By producing a hydropathy plot from the translated ORF, a prediction of whether or not DNA-binding protein is an integral membrane protein could be made. An antigenicity plot was also constructed, allowing assessment of which portions of the protein could be used to generate a monoclonal antibody. The MacDNAsis program predicted the secondary structure of DNA-binding protein based on the amino acid sequence. This structure was compared to the RasMol image of the protein, which also demonstrates tertiary structure and was obtained from the National Center for Biotechnology Information. A multiple sequence alignment was performed on the amino acid sequence of the protein in five different species, and a phylogenetic tree was produced to asses the degree of amino acid conservation between these species over time.
The Open Reading Frame (ORF)--selection and amino acid translation
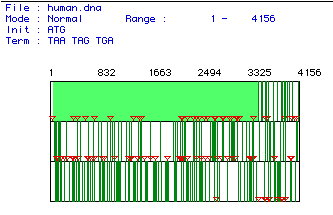
The human cDNA for DNA-binding protein was analyzed to determine all of the ORFs in the sequence. Figure 1 shows the results of that analysis. From this figure, it was determined that the largest ORF begins at neucleotide number 46 and terminates at neucleotide 3489. This is the first open reading frame, and it is highlighted in green in the figure below. This DNA segment constitutes a majority of the 4156 base pair sequence, and is the portion of the sequence used in further analyses.
Figure 1. Open reading frames in human cDNA. This figure shows all reading frames in the human cDNA for DNA-binding protein. The red triangles indicate initiation codons and the vertical green lines mark the stop codons. The neucleotide sequences of these codons aare listed--ATG for start codons; TAA, TAG, and TGA for stop codons. Neucleotide numbers are given above the reading frames, and the range of this analysis includes neucleotides 1-4156, the entire cDNA sequence of DNA-binding protein. The light green highlighted area marks the largest ORF, ranging from neucleotide 46-3489.
The largest open reading frame was translated by MacDNAsis into the amino acid sequence. Knowledge of the amino acid content of the sequence permitted the prediction of its molecular weight--128,275.80 daltons (128.3 kDa).
The Hydropathy Plot
Using a procedure developed by Kyte and Doolittle, a hydropathy plot was constructed from the amino acid sequence obtained above (Figure 2).
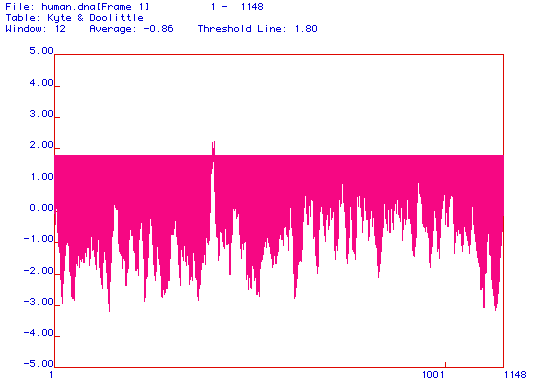
Figure 2. Hydropathy Plot. Consrtucted using the Kyte and Doolittle method, this plot illustrates the hydrophobicity of the protein. Posative values on the graph indcate hydrophobic regions, while negative areas show hydrophyllic portions of the protein. The axis of the plot is set at 1.8. A reading that exceeds a value of 1.8 indicates that the protein may have a transmembrane domain in that region. One peak in this protein cosses the axis (approximately 400aa) indicating that there may be one transmembrane domain. Amino acid numbers are designated across the bottom of the plot, and the range of amino acids is 1-1148.
A hydropathy plot graphically illustrates which areas of the protein are hydrophyllic (negative reading on the graph) and which are hydrophobic (posative reading). Proteins that yield mostly negative readings are not associated with cell membranes because they are not hydrophobic enough to cross the phospho-lipid bi-layer. Such proteins are often soluble and may be found in the cell cytoplasm.
In order to have a transmembrane domain, a segment of the protein must have a hydropathy reading greater than +1.8. This value defines hydrophobicity that is substantial enough to cross the phospholipid bi-layer. Figure 2 shows that DNA binding protein has one segment that crosses the 1.8 threshold. This suggests that this segment of the protein may have a transmembrane domain. However, this is the only peak in the plot that is hydrophobic enough to be a transmembrane domain, indicating that DNA-binding protein is most likelly not an integral membrane protein.
Antigenicity Plot
The amino acid sequence of the largest ORF was also used to produce and antigenicity plot (Figure 3).
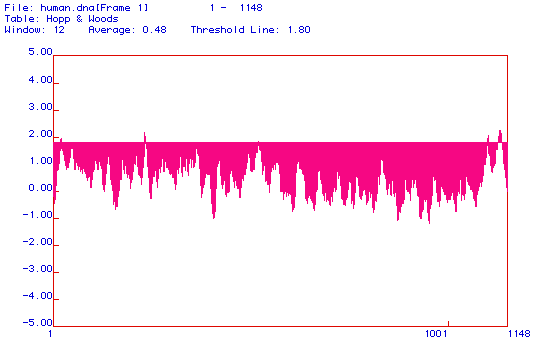
Figure 3. Hopp and Woods antigenicity plot. This plot shows the hydrophyllicity of this segment of protein. Posative values indicate hydrophyllic areas,which are also antigenic. The threshold line is set at 1.8, and the majority of the protein is located between this value and 0.00. The average hydrophyllicity of the area is given as 0.048. Amino acids 1-1148 are included in the range of the plot.
This model, developed by Hopp and Woods, shows the hydrophyllicity of the protein segment analyzed. Popsative values indicate hydrophyllic areas of the protein, which can not be associated with the phospho-lipid bi-layer. Figure 3 demonstrates that DNA binding protein is more hydrophyllic than hydrophobic, with an average value of 0.48.
Because hydrophyllic regions of the protein cannot be associated with the phospho-lipid bi-layer, they point away from the membrane into the cytoplasm and can therefore interact with antibodies in the cell. Monoclonal antibodies can be made against any region of the protein that has a large portion in the cytoplasm. As such, proteins that are very hydrophyllic are also very antigenic. Figure 3 thus shows that DNA-binding protein is not only hydrophyllic, but also antigenic. Monoclonal antibodies could be made against numberous areas of the protein, wherever it is substantially hydroophyllic.
Secondary Structure
The amino acid sequence obtained from the ORF cDNA was also used to predict the secondary structure of the protein (Figure 4). The secondary structure of a protein refers to the coiling and folding of the polypeptide chain in regular patterns, that results from hydrogen bonds. Examples of secondary structure are the beta-pleated sheet, in which teh chain folds back and forth, and the alpha-helix.
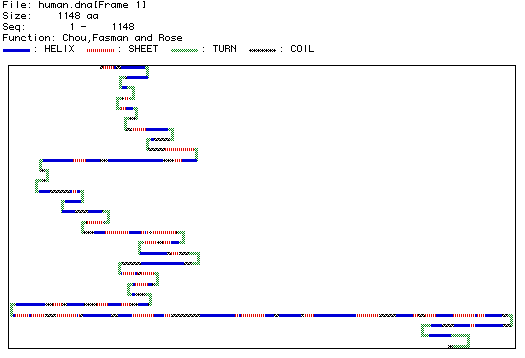
Figure 4. Image of secondary structure. This image was synthesized by MacDNAsis based on the amino acid sequence of the protein. It predicts the folds in the protein due to hydrogen bonds along the polypeptide backbone. Blue segments represent an alpha-helix, red shows the beta-pleated sheet areas, gray areas represent coils in the protein, and green areas represent the turns. This is an analysis of 1148 amino acids of human DNA binding protein.
This secondary structure predicts a protein segment with many turns, helical, and pleated areas. The predicted structure can be compared with a three-dimensional RasMol image of the actual protein, keeping in mind that figure 4 only displays part of the DNA-binding protein structure, while the RasMol image displays the whole protein with tertiary structure. Tertiary structure results from irregular bonding between side groups of the protein's amino acids.
To facilitate comparisons between the secondary structure of DNA-binding protein and its RasMol image, go to the menu bar at the top of the RasMol page. Click on "Display", and select "Ribbons". This allows you to view the protein's alpha helixes and pleated sheets more clearly. Next Click on "Colors" and select "structure". This option highlights the helixes is red and the pleated sheets in yellow. Although it is not possible to pick out the exact segment of protein whose secondary structure is predicted in Figure 4, similarities between the two can be noted. A series of many helixes and pleated sheets without any turns appears in the secondary structure. In the RasMol image, this segment is folded many times as a result of the side group bonding characteristic of tertiary structure.
Multiple Sequence Alignment
The amino acid sequence of DNA-binding protein was compared across the following five different species: C. elegans (Nematode), Drosophila (Fruit Fly), Homo sapiens (Human), Mus musculus (Mouse), and S. cervissae (Yeast). Figure 5 displays a portion of those amino acid sequences--from amino acid 851 to 1100. Click on the names of the species to view their entire amino acid sequences, as obtained from a search of the Genbank at NCBI.
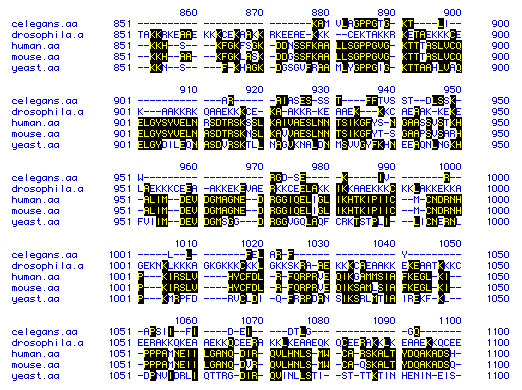
Figure 5. Multiple Sequence Alignment. The amino acid sequences of DNA binding protein in a nematode, a fruit fly, a human, a mouse and yeast are compared from amino acid 851-1000. The one-letter amino acid abbreviations are used. Yellow letters highlighted in black indicate amino acids that are present in more than one of the species (homologous). Dashes were added to improve the sequence alignment. Amino acid numbers are shown on the sides and at the top of each row of comparisons.
Although the sequences are obviously homologous in some areas and across many of the species, the dashes show that the sequences do not align naturally and that strict amino acid conservation is thus low.
The Phylogenetic Tree
A phylogenetic tree was constructed using the protein sequences of the five different species listed above (Figure 6).
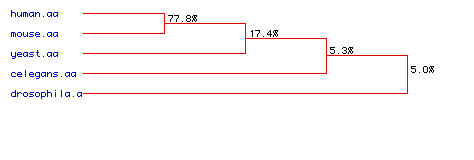
Figure 6. Phylogenetic Tree. This figure diagrams the degree of amino acid conservation over time across the given five species. The percentages given estimate the probability that the amino acid sequences evolved at the same time.
Examining the similarity of amino acid sequences of the same protein across different species allows analysis of amino acid conservation over time. Knowledge of amino acid conservation allows estimation of evolutionary relationships between species. From the figure we see that the human and mouse amino acid sequences have a higher degree of similarity and thus a higher probability that the DNA-binding protein sequence derived from the same origin. This would be expected since humans and mice are members of the same class, Mammalia.
To view the complete amino acid sequences of the species diagrammed in Figure 6, click on their names below:
Mouse (Mus musculus)
Fruit Fly (Drosophila)
Nematode (C. elegans)
Yeast (S. cervissae)
Human (Homo sapiens)
Binding it all together
The MacDNAsis analyses utilized the cDNA sequence of the human DNA-binding protein to obtain the amino acid sequence of the largest open reading frame. From this amino acid sequence analyses of protein structure, molecular weight, transmembrane domains and antigenicity were made. As well, comparisons between amino acid sequences of the protein in five species permitted estimation of homology and evolutionary relationships. These analyses underscore the possibilities for understanding protein structure, function, location, and evolutionary overlap that are obtained from neucleotide and amino acid sequences.