MacDNAsis analysis of triosephosphate isomerase sequences
from genome organisms
After collecting the cDNA and amino acid sequences for the five genome
organisms from Genbank (see this page for details),
I used the human cDNA sequence for analysis with the MacDNAsis sequence
analysis software. The results for the analysis are presented below.
Open Reading Frame (ORF) search
MacDNAsis was used to search the human cDNA sequence for open reading frames.
The search showed that the largest open reading frame was in the frame
starting with the second base pair in the cDNa sequence, and was from base
pair 263 to base pair 1117. The search results are shown in Fig. 1 below.
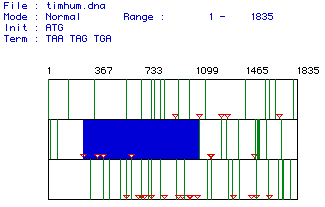
Figure 1. Search results for open reading frames using the MacDNAsis
program.Standard start and stop codons were used for the search. The red
inverted triangles represent start codons and the vertical green lines
represent the stop codons. The blue area marks the largest open reading
frame found, from base pair 263 to base pair 1117.
Protein sequence and molecular weight
The MacDNAsis program was used to translate the largest open reading frame
in the cDNA (see above) into the corresponding amino acid sequence, and
calculate the number of occurrences of each amino acid and the molecular
weight of the the protein. The results of the translation are shown in
Figure 2 below.
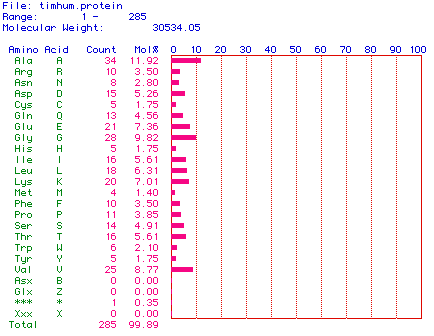
Figure 2. Amino acid content of the protein sequence translated from
the cDNA sequence. Note the molecular weight calculation of the protein.
The molecular weight of the protein sequence was calculated to be 30534.05
daltons, or about 30.5 kDa.
Hydropathy plot of amino acid sequence
A hydropathy plot for the translated protein sequence was done using the
Kyte and Doolittle algorithm. The results are shown in Fig.3.
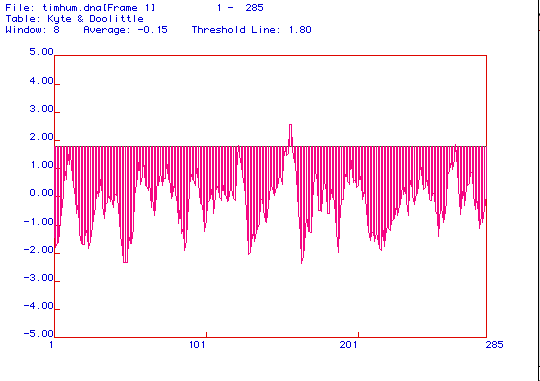
Figure 3. Hydropathy plot for protein sequence (translated from human
triosephosphate isomerase cDNA) using the Kyte and Doolittle algorithm.
A window of 8 amino acids was used and the threshold for a transmembrane
domain was 1.8.
The hydropathy plot shows that there is only one peak that reaches the
threshold of 1.8 used to determine the existence of a transmembrane domain.
Triosephosphate isomerase is involved in glycolysis which takes place in
the cytoplasm of a cell, and should not be a integral membrane protein.
Therefore the region of high hydrophobicity is probably just one that is
on the inner side of the tertiary structure, and does not really indicate
a transmembrane region. We must remember that the Kyte and Doolittle algorithm
is merely a computer prediction of tertiary structure, and may not be correct.
Antigenicity analysis
An antigenicity plot was generated for the translated protein sequence
using the Hopp and Woods algorithm. The plot is shown in Fig. 4.
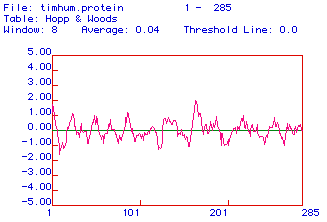
Figure 4. Antigenicity plot for protein sequence (translated from human
triosephosphate isomerase cDNA). The algorithm used was that of Hopp and
Woods and the window used was 8 amino acids.
The antigenicity plot is an indication of the areas of the protein that
are hydrophilic and highly charged, making it more likely that these regions
would be on the outside of a tertiary structure. Such charged regions of
the protein would be the ones most easily used as epitopes for antibodies
the protein. For our protein sequence we see that there are several promising
regions, but the best choices are either the N-terminus or a region between
the amino acids 165-180. These regions are most likely to be "sticking
out" out of the protein tertiary structure, and would be those best accessible
to an antibody molecule.
Secondary structure prediction
I used the Chou, Fasman and Rose algorithm to predict the secondary structure
of the protein using the primary sequence. The results are shown in Fig.5.

Figure 5. Secondary structure prediction for protein sequence translated
from human triosephosphate isomerase cDNA. The algorithm used was that
of Chou, Fasman and Rose. The "H" strings mark helical structure, the "S"
strings mark sheets, the "t"s mark turns and the "C"s mark coils.
The secondary structure predicted is typical of a globular protein,
with a good mixture of helical coils and pleated sheets. This is expected
for an enzyme like triosephosphate isomerase and can be seen in the Rasmol
image of triosephosphate isomerase (MMDB Id: 2490, PDB Id: 1YPI).
Multiple sequence alignment and phylogenetic tree
The Genbank amino acid sequences for triosephosphate isomerase from the
five genome organisms was used to perform a multiple sequence alignment
test for sequence homology. The original protein sequences can be found
here - human, mouse,
Drosophila, yeast
and C. elegans. The results of the alignment
are shown in Fig.6.

Figure 6. Multiple alignment results for the amino acid sequences for
triosephosphate isomerase from the five genome organisms - human
(timhum.aa), yeast (timsac.aa), mouse
(timmus.aa), Drosophila (timdro.aa) and
C.elegans (timcel.aa). The consensus
sequences are highlighted in black.
The multiple sequence alignment results show that a large proportion
of the amino acids in the primary sequence of triosephosphate isomerase
match up. This probably indicates that this protein has been highly conserved
through evoultion, since it plays such an important role in metabolism.
I also used the MacDNAsis program to generate a phylogenetic tree based
on the sequence homology of triosephosphate isomerase. The tree is shown
in Fig. 7.
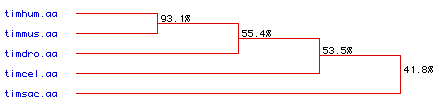
Figure 7. Phylogenetic tree based on the sequence homology of triosephosphate
isomerase for the five genome organisms - human
(timhum.aa), yeast (timsac.aa), mouse
(timmus.aa), Drosophila (timdro.aa) and
C.elegans (timcel.aa). The percentage
homology with the human amino acid sequence is indicated on each branching
point.
[Top] [Home] [Back
to the Davidson College Molecular Biology page]
Comments? Questions? Suggestions? E-mail rakarnik@davidson.edu.