This web page was produced as an assigment for an undergraduate course at Davidson College.
Phenylalanine Hydroxylase
The Protein
Phenylalanine hydroxylase (PAH) is an enzyme which is used in the conversion of ingested phenylalanine to tyrosine, as is illustrated in Figure 1. Tyrosine differs from phenylalanine with the addition of a hydroxyl group to the phenyl ring - this addition is accomplished by phenylalanine hydroxylase, and does not occur without it. PAH is active in the liver and is part of the metabolic process involving the processing of ingested amino acids. After being processed, these amino acids move from the liver to the bloodstream, where they are available to cells that are making proteins (Purves et al., 2001; Dolan DNA Laboratory, 2002). PAH is also found in the species Mus musculus and Rattus norvegicus (mice and rats), and may be found in additional organisms as well (NCBI, 2003).
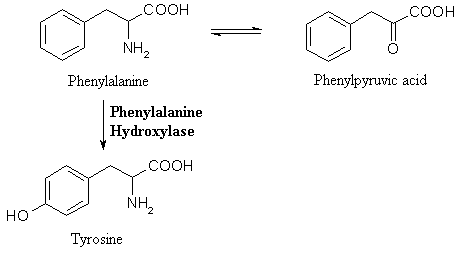
Figure 1: A Partial Representation of the Metabolic Pathway of Phenylalanine. These equations show the first step in the breakdown of phenylalanine within the body. Phenylalanine is in equilibrium with phenylpyruvic acid, but when phenylalanine hydroxylase is present, phenylalanine is converted into tyrosine through the catalytic addition of a hydroxyl group. If PAH was not present within a body, phenylalanine would not be converted to tyrosine and would merely remain in the body, meaning that the concentrations of both phenylalanine and phenylpyruvic acid would increase as more phenylalanine is ingested. The chemical equations and structures for this figure were found in Purves, 2001. The structures were drawn for this website using ISISDraw 2.4.
Phenylketonuria - PAH-Related Disease
Mutations within the PAH protein in humans result from any number of mutations within the PAH gene on Chromosome 12. A total of 400 disease causing mutations of the PAH gene have been discovered (Ryan et al., 2003). The extent of mutation can have different effects on the breakdown of phenylalanine. Mutations can cause the substrate of the PAH enzyme to change so that it no longer recognizes phenylalanine, the speed at which PAH catalyzes the breakdown of phenylalanine is changed, or the entire PAH enzyme may be broken down (Dolan DNA Laboratory, 2002).
If PAH is not produced or if it has mutated, phenylketonuria (PKU), non-PKU hyperphenylalaninemia (non-PKU HPA), and variant PKU can be the result. Sufferers of these diseases have elevated levels of phenylalanine in their blood plasma, since it can not be broken down without PAH. These three diseases range in severity with PKU being the most severe, as it occurs when almost no PAH is active. Non-PKU HPA is the least severe of these three disorders. Blood obtained from heelpricks performed on newborns is tested for these disorders through the Guthrie Microbial Assay. This assay tests the concentration of phenylalanine in plasma - a normal concentration is 120 micromols per liter of plasma. For a person with PKU, the concentration of phenylalanine is above 1000 micromols per liter of plasma, and non-PKU HPA sufferers have plasma phenylalanine concentrations between 120 and 1000 micromols per liter (Ryan et al., 2003).
Untreated PKU results in irreversible mental retardation due to a lack of cognitive development. While no definite mechanism for how PKU affects brain development has been suggested, there is evidence that phenylpyruvic acid might inhibit pyruvate carboxylase in the brain, which may cause a defect in the production of myelin, which in turn causes mental retardation (McKusik, 1966-2003). People with PKU have lower levels of the neurotransmitter, dopamine, than people who do not have PKU, which has led to some hypothesizing that this could contribute to mental retardation (Dolan DNA Laboratory, 2002).
Other less significant symptoms of PKU include a "mousy" or "musty" odor which occurs when phenylalanine is excreted. Also, those people who have PKU are sometimes lighter in pigmentation, have unusual body postures, eczema and epilepsy (McKusik, 1966-2003).
Treatment
Phenylketonuria is completely treatable using dietary restriction. If a person who has PKU does not ingest phenylalanine, that person is unable to build up to dangerously high concentrations within his or her blood. For this reason, all newborns in most developed countries are tested for PKU within a couple of weeks of birth, so that dietary restrictions can be imposed on people with PKU before permanent brain damage occurs. Automatic PKU testing began in the 1960's in America, and spread to other developed nations in the early 1970's (Ryan et al., 2003).
In addition to a modified diet, PKU is treated with supplemental BH4 which has been shown to lower phenylalanine concentrations in the plasma of some patients. BH4 is a cofactor in the breakdown of phenylalanine, and so some mutated PAH enzymes are thought to be less able to bind to BH4, and therefore are less able to break down phenylalanaine. The addition of supplemental BH4 will saturate the binding sites, even with a lowered binding affinity, creating more active PAH enzymes (Ryan et al., 2003).
Inheritence of Disease
Phenylketonuria is a genetic disorder, and is autosomal recessive. This means that if both parents are heterozygous for the PKU mutation on the PAH gene, their children have a 25% chance of being homozygous for PKU and expressing the disease, and a 50% chance of being heterozygous carriers themselves. If one parent has PKU (meaning that it is homozygous for the recessive allele), then his or her children would have a 100% chance of being carriers, and a 50% chance of being affected by the disease (Ryan et al., 2003).
The children of women with PKU can be born with congenital heart disease and have impaired cognitive ability. There is evidence that if a woman has a sufficiently controlled diet during and preceding pregnancy her children will not suffer any damage, but even so, the risks of poor fetal or child health are much greater in women with PKU, and in some studies, all children studied suffered some effects - either developmental or behavioral. In one study of women whose plasma phenylalanine levels differed during pregnancy, women with low levels of phenylalanine had no incidence of congenital heart disease in their children, and lower incidences of microcephaly (6%) while in women with higher levels of phenylalanine, 14% of babies had congenital heart disease and 85% of children had microcephaly (Ryan et al., 2003; McKusik, 1966-2003).
The prevalence of PKU differs between races, from a prevalence of 1/26 in the Turkish population to 1/225 in the Finnish or Ashkenazi Jewish population (Ryan et al., 2003).
References
Dolan DNA Learning Center. 2002. Your Genes, Your Health: A Multimedia Guide to Genetic Disorders: Phenylketonuria Video. Cold Spring Harbor Laboratory. <http://www.yourgenesyourhealth.org/ygyh/mason/ygyh.html?syndrome=pku§ion=whatisit&video=0> Accessed 2003 March 13.
Heller HC, Orians GH, Purves WK, Sadava D. 2001. Life: The Science of Biology. 6th ed. Sunderland (MA): Sinauer Associates, Inc.; p 251, p 331-333
Ryan S, Scriver CR. 2003 Jan 24. Phenylalanine Hydroxylase Deficiency. Gene Reviews. <http://www.geneclinics.org/profiles/pku/details.html> Accessed 2003 March 13.
McKusik VA. 1966-2003. Phenylketonuria (#261600). <http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=261600> Accessed on OMIM database, 2003 March 13.
National Center for Biotechnology Information (NCBI), 2003. Entrez: Genomes - searchable Genetic Maps of Various Organisms. <http://www.ncbi.nlm.nih.gov/mapview/map_search.cgi?chr=mouse_chr.inf&query=PAH&qchr=&strain=&neighb=off> <http://www.ncbi.nlm.nih.gov/mapview/map_search.cgi?chr=rat.inf&query=PAH&qchr=&neighb=off>
Send questions, comments, and suggestions to Jamie Causey.