This web page was produced as an assignment for an undergraduate course at Davidson College
HEXA Gene & Beta-hexosaminidase A Enzyme
Location
The enzyme Beta-hexosaminidase A is found in the lysosomes of cells (NCBI,
2003). Lysosomes degrade large molecules into smaller monomers that the cell
can use. When beta-hexosaminidases are mutated some of the biggest problems
can be seen in the neuronal lysosomes which affect the nervous system (Medical
Neurobiology, 2003). These changes have drastic effects that appear as symptoms
of diseases such as Tay-Sachs.
Figure 1. These three images show different views of the enzyme beta-hexosaminidase A. Images from PDBsum.
Function
The enzyme beta-hexosaminidase A is comprised of an alpha-subunit and a beta-subunit.
The alpha-subunit is encoded for in a gene called HEXA found on chromosome
15 (OMIM,2001). Both subunits are required for proper beta-hexosaminidase
A functioning and binding. Beta-hexosaminidase A is one part of a complex
that degrades the lipid GM2 ganglioside. It binds to another molecule called
GM2 activator protein to form this complex. With all parts functioning normally
this complex hydrolyzes GM2 ganglioside. It does this by removing a beta-1,4-linked
N-acetylhexosamine residue from the GM2 ganglioside (Medical Neurobiology,
2003). By removing this residue from GM2 ganglioside, the complex transforms
it into GM3 ganglioside. The alpha-subunit is very important in the degradation
process of GM2 ganglioside into GM3 ganglioside because the alpha-subunit
is the specific region that hydrolyzes the lipid (OMIM,2001). The beta-subunit
region is not capable of this specific action. But even with these functional
differences both subunits are vital to the function of beta-hexosaminidase.
If one subunit is malfunctioning, beta-hexosaminidase cannot form the hydrolyzing
complex with GM2 activator protein (NCBI, 2003).
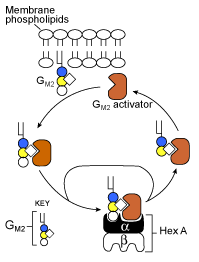
Figure 2. Shows the mechanism for GM2 ganglioside degradation by beta-hexosaminidase A. The GM2 activator protein binds to GM2 ganglioside which is attached to the lysosome's membrane phospholipids. It then forms a complex with beta-hexosaminidase A by binding to the alpha-subunit. Once this complex is formed, it can hydrolyze the beta-1,4-linked N-acetylhexosamine residue transforming it into GM3 ganglioside. After this is complete the GM2 activator protein releases the newly formed GM3 ganglioside. Image from NCBI.
Mutants
HEXA gene:
The most common mutation of the HEXA gene resulting in Tay-Sachs disease is
a four base pair insertion found in exon eleven (OMIM,2001). Another fairly
common mutation is a splice junction mutation found at intron twelve (Navon,
1989). There are many mutations of the HEXA gene that result in Tay-Sachs
and many that seem to have no affect.
Beta-hexosaminidase A:
Mutations of the HEXA gene can cause the alpha-subunit of beta-hexosaminidase
A to malfunction or not be produced at all. These mutations are a recessive
trait passed down from parents to offspring. For the mutations to cause harm
both chromosomes must carry the recessive trait. When no alpha-subunit is
produced, no hydrolyzing complex can form with GM2 activator protein and thus
GM2 ganglioside cannot be hydrolyzed into GM3 ganglioside. If the alpha-subunit
is formed but is somehow different from the wildtype alpha-subunit version
due to a mutation of HEXA, beta-hexosaminidase A also is unable to bind to
GM2 activator protein and thus is unable to degrade GM2 ganglioside to GM3
ganglioside. Because GM2 ganglioside cannot be broken down, it accumulates
in the lysosomes (NCBI, 2003). These abnormal lysosomes filled with GM2 ganglioside
accumulate in the cell and can block other cellular processes. If this continues
the cell is eventually killed and destroyed. In the nervous system, neurons
with GM2 ganglioside can build up and cause serious damage. This can be seen
in the symptom’s of individuals suffering from Tay-Sachs disease. The
accumulation of GM2 gangliosides in neurons is so drastic that the brains
of Tay-Sachs individuals living for more than one year can increase in weight
by more than 50% (Medical Neurobiology, 2003). Symptoms of Tay-Sachs disease
include blindness, loss of motor function, paralysis and psychosis. In most
cases, patients don’t survive past early childhood (Navon, 1989).

Figure 3. This image shows a child with Tay-Sachs disease resulting from a mutation of the HEXA gene on chromosome 15. Often times individuals with Tay-Sachs disease look normal until the sudden onset of symptoms. Image taken from Loving and Losing a Child. Permission pending.
Chime Image of beta-hexosaminidase A-
Click
here!
Send comments, questions, and suggestions to: keholmberg@davidson.edu