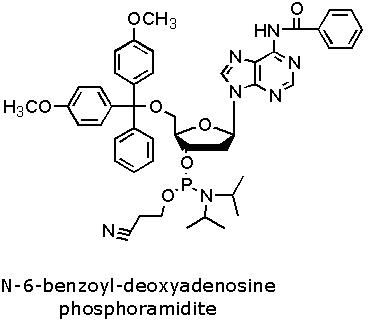
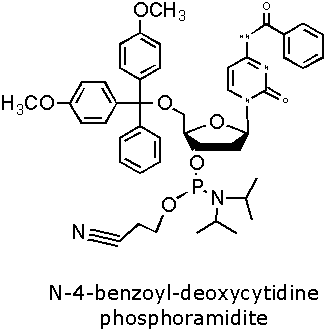
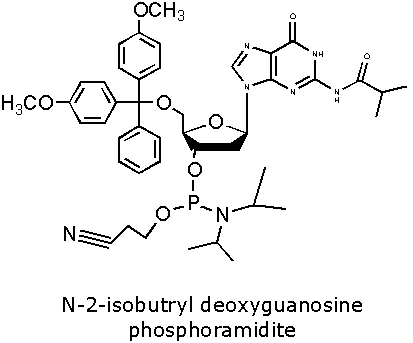
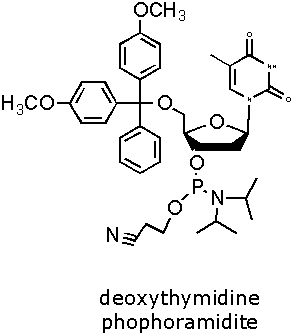
Figure 2. DNA Phosphoramidite Monomer Bases. Image used with
permission from Integrated DNA Technologies.
Step 1: De-blocking
The first base, which is attached to the solid support, is at
first inactive because all the active sites have been blocked or protected.
To add the next base, the DMT group protecting the 5'-hydroxyl group must
be removed. This is done by adding a base, either dichloroacetic acid (DCA)
or trichloroacetic acid in dichloromethane (DCM), to the reaction column (IDT,
2000). The 5’-hydroxyl group is now the only reactive group on the base
monomer. This ensures that the addition of the next base will only bind to
that site. The reaction column is then washed to remove any extra acid and
by-products (BioSource, 2003).
Step 2: Base Condensation
The next base monomer cannot be added until it has been activated.
This is achieved by adding tetrazole to the base. Tetrazole cleaves off one
of the groups protecting the phosphorus linkage (IDT, 2000). This base is
then added to the reaction column. The active 5’-hydroxyl group of the
preceeding base and the newly activated phosphorus bind to loosely join the
two bases together. This forms an unstable phosphite linkage. The reaction
column is then washed to remove any extra tetrazole, unbound base and by-products
(BioSource, 2003).
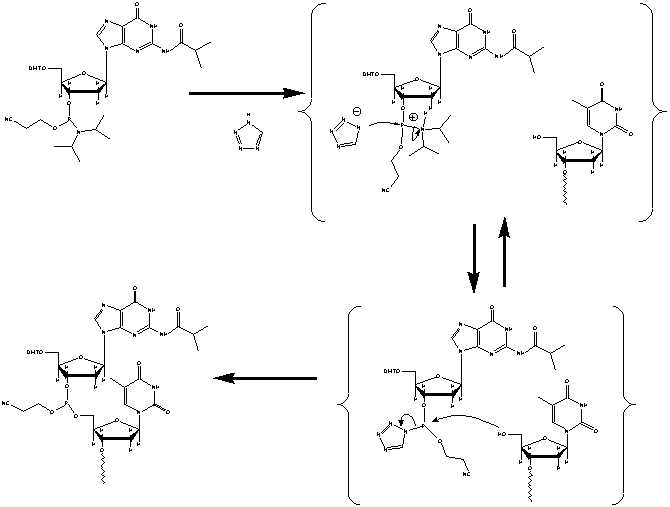
Figure 3. Top left: A fully blocked base next in line to be
added onto previous base. Top Right: The next base is now activated by tetrazole
(left) and the previous base is de-blocked ready for a base addition (right).
Bottom Right: 5'-hydroxyl of previous base binds to phosphorus linkage of
next base (arrow). Bottom Left: The base has been successfully been added
to the previous base. The Image used with permission from Integrated
DNA Technologies.
Step 3: Capping
When the activated base is added to the reaction column some
does not bind to the active 5’-hydroxyl site of the previous base. If
this group is left unreacted in a step it is possible for it to react in later
additions of different bases. This would result in an oligonucleotide with
a deletion. To prevent this from occurring, the unbound, active 5’-hydroxyl
group is capped with a protective group which subsequently prohibits that
strand from growing again. This is done by adding acetic anhydride and N-methylimidazole
to the reaction column (IDT, 2000). These compounds only react with the 5’-hydroxyl
group. The base is capped by undergoing acetylation. The reaction column is
then washed to remove any extra acetic anhydride or N-methylimidazole (BioSource,
2003).
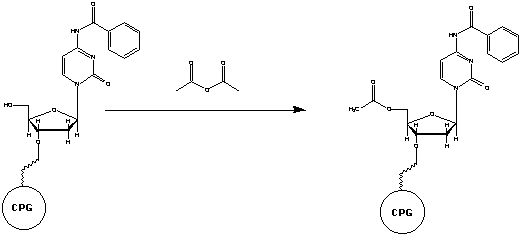
Figure 4. The base on the left (already attached to the solid
support) did not bind to a base in the Base Condensation step. The unreacted
5'-hydroxyl is blocked from further reactions by acetylation. Image used with
permission from Integrated DNA Technologies.
Step 4: Oxidation
In step 2 the next desired base was added to the previous base,
which resulted in a unstable phosphite linkage. To stabalize this linkage
a solution of dilute iodine in water, pyridine, and tetrahydrofuran is added
to the reaction column (IDT, 2000). The unstable phosphite linkage is oxidized
to form a much more stable phosphate linkage.
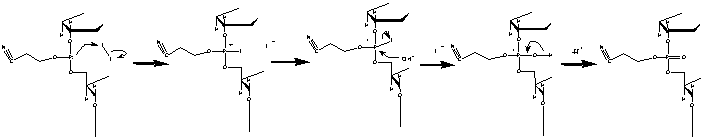
Figure 5. Illustrates the oxidation process of the unstable
phophite linkage to a more stable phosphate linkage. Image used with permission
from Integrated DNA Technologies.
Repeat
Steps one through four are repeated until all desired bases
have been added to the oligonucleotide. Each cycle is approximately 98/99%
efficient (IDT, 2000).
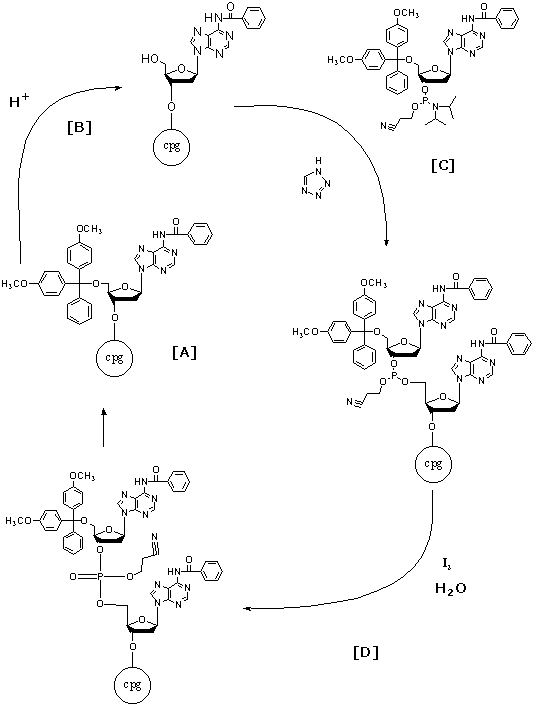
Figure 6. The complete synthesis cycle including de-blocking
(B), base condensation (C), capping (C), and oxidation (D)are illustrated.
This cycle is completed once for each additional base desired. Image used
with permission from Integrated DNA Technologies.
Post Synthesis
After all bases have been added the oligonucletide must be cleaved
from the solid support and deprotected before it can be effectively used.
This is done by incubating the chain in concentrated ammonia at a high temperature
for an extended amount of time. All the protecting groups are now cleaved,
including the cyanoethyl group, the heterocyclic protection groups, and the
DMT group on the very last base (BioSource, 2003).
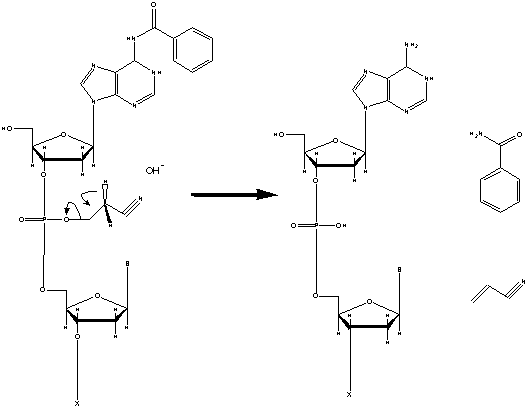
Figure 7. The group protecting the heterocyclic primary amine
and the cyanoethyl are both cleaved with concentrated ammonia. Image used
with permission from Integrated DNA Technologies.
Final Product
The final product is a mixture of the sought after oligonucleotide,
cleaved protective groups and oligonucleotides with internal deletions (IDT,
2000). This mixture of wanted and unwanted species is what the oligonucleotide
synthesis companies send to the recipient. To obtain a solution only containing
the desired oligonucleotide, the recipient must desalt the heterogeneous mixture.
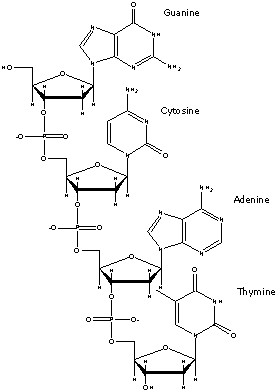
Figure 8. An example of a oligonucleotide. Image used with
permission from Integrated DNA Technologies.
Desalting
Desalting is done to purify the solution. It removes any species
that may interfer with future reactions. The major problematic ingredient
in the heterogeneous mixture is the ammonium ion. To filter the solution of
the ammonium ions three different methods can be utilized. They are ethanol
precipitation, size-exlusion chromatography, or reverse-phase chromatography
(BioSource, 2003).

Figure 9. A example of a PAGE purification method. The oligonucleotides desired
from this process are the dark blots. The oligonucleotides with internal deletions
and the leftover protecting groups are not visible, but labeled as Unwanted
Truncation Products. Image used with permission from Integrated
DNA Technologies.
References:
BioSource International. 2003 Feb 10. BioSource home page. <http://www.biosource.com>.
Accessed 2003 Feb 11.
Integrated DNA Technolgies. Copywrite 2000. IDT home page. <http:.//www.idtdna.com>.
Accessed 2003 Feb 11.
Purves, W., Sadava, D., Orians, G., Heller, H. 2001. Life: The
Science of Biology. 6th ed. Sinauer Associates, Inc. p. 210-217.
Back
to Homepage