Favorite Gene/Protein
*This website was produced as an assignment for an undergraduate course at
Davidson College*
Tropomyosin
Starting with Some Basics of Muscles:
Skeletal muscle is composed of cells, called fibers, that are
specialized to contract or shorten in length. Each fiber is made of smaller
subunits called myofibrils that are composed of contractile proteins called
myosin and actin which are responsible for muscle contraction at the molecular
level (Purves et al. 2001). These contractile protein filaments are
also called thick (myosin) and thin (actin) filaments. Here the focus is on
muscle fibers and the protein consituents thereof. The fibers of muscle are
made up of a number of different proteins arranged in a very specific manner
(Carlson 1974).
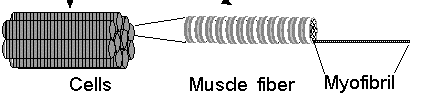
Figure 1. The general breakdown of muscles into sub-structures.(Permission
Pending from Ohio State University)
Muscle contains approximately 70 to 75% water and 20 to 22 %
protein. The protein component can be divided into the myofibrillar proteins
(salt soluble), which make up about 50 to 55% of the total and are responsible
for the actual contraction of the muscle, and the sacroplasmic or soluble
proteins that make up about 25 to 35% of the total (Stracher 1983). These
proteins are almost all enzymes. Focusing on the more abundant contractile
proteins, fibers need to be the initial discussion point. Muscle fibers consist
of thick (myosin) and thin (actin) filaments, as mentioned earlier. Two other
proteins associate with actin to form the thin filaments, known as tropomyosin
and troponin proteins. (Together myosin, actin, tropomyosin, and troponin
make up over three-quarters of the protein in muscle fibers (Kimball 2003)).
5 to 8% of the thin filaments consist of the protein called tropomyosin (Kimball
2003).
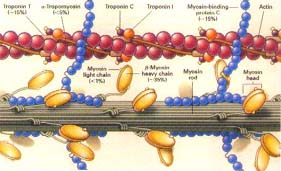
Figure 2. This image shows most of the components of
muscle
cells just to provide a general overview while further detail
will be drawn to a specific protein, tropomyosin, in this figure. (Permission
Pending)
Tropomyosin
A researcher, by the name of Bailey, first identified and
isolated Tropomyosin (1946) because its behavior in solution caught his attention.
It appeared to him that there was a protein with properties opposite that
of actin (Maciver 2003). Further research has shown that tropomyosin proteins
(abbreviated as TM) are found in virtually all eukaryotic cells in association
with actin and have similiar solubility and structural properties as those
of myosin, hence its name.(Barany 2002). TM molecules exist as dimers of two
identical a-helical polypeptide strands arranged
in a parallel, coiled-coil configuration. This rod-shaped molecule is typically
about 400 Å long and 20 Å wide with a molecular weight of 66 kD.
(Kreis and Vale, Ed. 1993). Each TM protein is bound or polymerized within
head-to-tail (with N and C termini on opposite ends of one strand) residues
via a manner of overlapping their ends of 8-11 amino acids. It was found that
eliminating 11 amino acid residues on the C-terminus eliminated polymerization
and markedly reduced the cooperative binding that tropomyosin regularly does
with actin. In conjunction with troponin (the other and more abundant actin-associated
protein; required presence for tropomyosin to bind to actin), the group of
TM proteins normally bind to the sides of an actin filament and regulate the
interaction of the filaments with myosin in response to Ca2+ (Baxevanis and
Ouellette, Ed. 1998). Removal of 10 residues at the N-terminus was seen to
virtually eliminates actin binding, just as occured with removal of amino
acids from the C-terminus. Such binding process is pivotal in the contractile
movement of skeletal and cardiac muscle cells, but actin binfing can be manipulated
through the addition of troponin. In properly functioning skeletal and cardiac
muscles, TM together with the troponin complex, serves as a component of the
regulatory mechanism by which the interaction of the F-actin (F denotes the
long-fiber form of actin) and myosin is controlled by the level of calcium
ions. In smooth muscle and nonmuscle cells in which the troponin complex is
absent, TM is still present, yet its role is less clear (Stracher 1983).
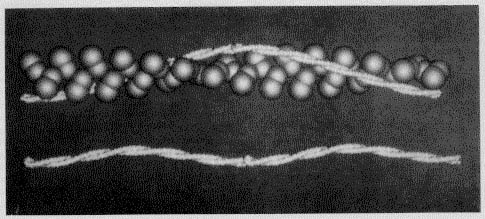
Figure 3. This image is used to show the binding interaction
of tropomyosin with seven actin actin monomers(shown in the upper strand)
and the head-to-tail binding of subsequent tropomyosin molecules to form two
coiled-coil strands (in the lower image). Permission
Pending from Dr Barany at the University of Illinois at Chicago).
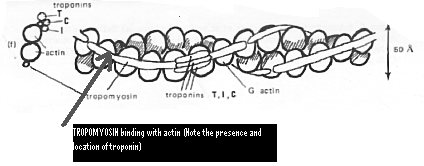
Figure 4. This image reveals another look at the binding
of tropomyosin with actin, in conjunction with troponin.
(Permission
Pending from Ohio State University)
Tropomyosin was one of the first actin-binding proteins to be
studied using immunofluorescence (Cooper 2002). This technique and several
others since then (including SDS-PAGE, X-ray analysis, etc.) have revealed
the two distinct strands of tropomyosin. The two strands are differentiated
by the terms a (fast) or b
(slow). The molecular weight of the chains does not differ significantly,
it is in the range of 33 kD-35 kD (Stracher 1983). Both chains have 284 amino
acid residues, but they differ in 39 of those. The amino acid sequence shows
a repeating pattern of nonpolar and polar residues, totaling of 7 residues.
The molecular length of a chain calculated from sequence, 284 x 1.49 Å
= 423 Å, (number of residues multiplied by the effective residue translation
in a coiled coil) is larger than the length of 400 Å calculated from
X-ray studies. This difference can be accounted for by assuming an overlap
of 8-9 residues between the ends of TM molecules.The ratio of the concentration
of the a and b subunit
varies with the muscle type. Slow (red) skeletal muscle and fetal muscle contain
a larger portion of the b subunit than fast (white)
skeletal muscle, whereas rabbit and avian cardiac muscle contain only the
a subunit. Speaking of other organisms than humans,
vertebrates have been known to express approximately 20 slighty different
tropomyosins, from alternate versions of four known tropomyosin genes (Maciver
2003). Two dimensional gel electrophoresis revealed several isoforms of TM
in different muscles. Furthermore, some of the TM isoforms can be phosphorylated.
Changes in the distribution of TM isoforms were observed during development
(Gordon, Regnier and Homsher 2001).
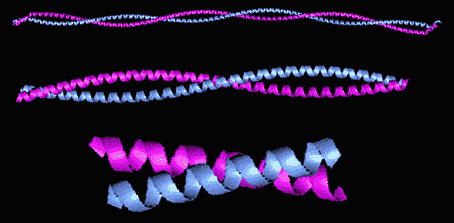
Figure 5. Distinctions Clear on this image of two separate
tropoyosin strands. Permission
Pending From Thomas Heimberg.
The Mechanics of Tropomyosin-Actin Binding:
Structural and Biochemical Basis
Contraction occurs when myosin S1 heads of the thick filament
attach to and exert force on actin molecules in the thin filament. This force
causes the thin filament to slide over the thick filament and the sarcomere
to shorten. The shortening of the sarcomere develops a force implying that
the muscle must move. Ca2+ binding to the troponin (Tn) on the thin filament
initiates the force-generating interaction of myosin and actin, and ATP hydrolysis
provides the energy for the molecular changes that drive force generation
and muscle shortening.
The basic structure is soo key to understand: helically-arranged
actins form the backbone of the thin filament, with the regulatory proteins
(tropomyosin (TM) and Troponin (Tn)) attached to actin (A) in a ratio of 7:1:1
(A:TM:Tn). TM binds to 7 actin monomers in the thin filament helix and overlaps
the adjacent TMs. Tn attaches to two actins in the absence of Ca2+ through
its TnI subunit and to TM through the TnT subunit at the TM/TM overlap zone.
Ca2+ binding to the TnC subunit strengthens the TnC-TnI interaction (interactions
that form the troponin complex mentioned earlier) and detaches TnI from its
contacts with actin. Recently structural studies have shown that this Ca2+-mediated
detachment of TnI from actin allows TM to move over the surface of the
actin filament. On the actin surface there are sights for weak (consisting
mainly of electrostatic) and strong myosin binding. TM either rolls around
its axis or slides from a position near the outer edge of the thin filament
(where it covers many of the myosin binding sites on the actin). Once it slides,
the actin and myosin can't bind, therefore the muscle relaxes. TM is a
flexible molecule and its positioning should be considered dynamic (Gordon,
Regnier, and Homsher 2001).Tropomyosin does not occupy a single fixed position
in the presence of elevated concentrations of Ca2+. Rather, the TM "rocks
and rolls" or "slips and slides" back and forth over the actin
surface. Therefore, the following linked movie, which shows Ca+ binding and
subsequent contractile events, should be viewed as an average position of
the tropomyosin. (Animation
of Muscle Contraction ~Permission Pending from Dr Campbell of Davidson
College). When the myosin is strongly bound to actin, TM is locally stablilized
in a position that makes both weak and strong myosin binding sites available
on nearby actin monomers. Thus, research has revealed that thin filament activation
is achieved by the movement of TM over the actin surface, which is controlled
both by Ca2+ binding to TnC and initial strong myosin binding to actin. This
TM binding allows for forced generation of movement and shortening.
Figure 6. Now it's Your Turn To Make the Tropomyosin
Dynamic using this Chime Model. Permission
Pending from Dr Rule at Carnegie Mellon University.
Options for Viewing: Rotate (Please note that this feature will be added at a later date.)
Studies have suggested that there are three states of the thin
filament:
 Absence of
Ca2+: resulting in TM blocking access to actin binding sites
Absence of
Ca2+: resulting in TM blocking access to actin binding sites
"Closed"
State: When myosin weakly binds to actin
"Open"
State: Myosin can strongly bind to actin
In the presence of Ca2+, the position of TM is typically such
that ~20% of the actin sites at any time are in the "open" state
and ~80% are in the "closed" state, which corresponds to an average
position of TM (Gordon, Regnier, and Homsher 2001). This breaks down to there
being a considerable correlation between the structural and biochemical states
of thin filament activation as defined by the TM position and the ability
of myosin to bind either weakly or strongly to actin.
The movement of TM is similiar, though not exactly the same
in skeletal (mentioned above) versus cardiac muscle. Both types of contractile
motion are activated rapidly by Ca2+ binding to TnC and the consequent movement
of TM. The difference is basically the extent of actin filament movement (larger
in skeletal muscles) (Cooper 2002).
In conclusion, when cardiac and skeletal muscle are in resting
positions, TM on the thin filament blocks strong binding of myosin to actin.
Ca2+ binding to Tn allows TM to move over the thin filament, exposing some
myosin binding sites. Strong attachment of myosin stabilizes the position
of TM proteins, which in turn allows for further strong myosin binding and
force development. Additionally, it is important to note that recent studies
have shown that other proteins can serve as tropomyosin-binding agents, in
place of troponin; such proteins include: calponin, endostatin, enigma, gelsolin
(sub-domain 2), S100A2 (Maciver 2003).
Tropomyosins and Human Diseases
Certain diseases have been attributed to alterations in tropomyosins.
For example: Mutations and deletions of tropomyosin genes and the altered
expression of such genes have been reported as associated with transformation
of vertebrate cells and further that over-expressed of normal tropomyosin
can inhibit transformation. So far research has indicated that in some cases
at least this is due to the 3' untranslated region of the gene (Cooper 2002).
The oncogene Ras has been implicated in tropomyosin mediated cell transformation
that has in turn been associated with actin cytoskeleton alteration.
Allergies such as asthma seem to be increasingly common problem
for no obvious reason. Many other allergies are caused by reactivity against
cytoskeletal proteins. An example of this is that many hay fever sufferers
produce a response to profilin from the pollen of many plants, which links
further back to tropomyosin. Tropomyosin has recently been connected to human
breast cancer, too. It turns out that human breast cancer cell lines and invasive
tumors frequently have reduced tropomyosin expression (Baxevanis and Ouellette
1998).
Hypertrophic Cardiomyopathy: Hypertrophic cardiomyopathy
(HCM) is usually an inherited disorder caused by mutations in a number of
cardiac cytoskeletal proteins including myosin II heavy chain, troponin T
and tropomyosin. This condition stems from the heart muscle thickening and
therein blood not pumped obtimally.
The emerging pattern is that HCM mutations increase sliding speed of tropomyosin,
increase Ca2+-sensitivity and/or impair relaxation i.e. a hypercontractile
phenotype. DCM mutations decrease sliding speed and Ca2+-sensitivity (Maron
2002). This translations into a entire host of heart conditions for people
with mutations in a regulatory protein, such as tropomyosin .
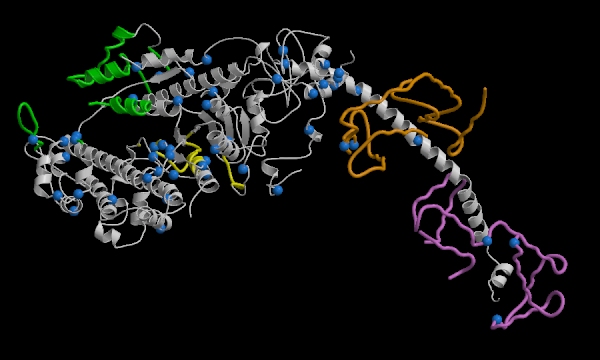
Figure 7. This image represents muscle fibers. The key element is
the blue dots which represent all the residues which when mutated translate
into hypertrophic cardiomyopathy. (Permission
Pending from Dr Watkins)
color key:
GREEN = actin-binding domain
YELLOW = ATP-binding site
ORANGE = essential light chain
VIOLET = regulatory light chain
BLUE = residues which, when mutated, cause hypertrophic cardiomyopathy
References
Barnay, M.; Barnay, K. Biochemistry of Muscle Contraction.
March 2002. The University of Illinois at Chicago. 12 March 2003.<http://www.uic.edu/classes/phyb/phyb516/regulatoryproteinsu.htm>
Baxevanis, Andreas; Ouellette, B.F. Francis.1998 Bioinformatics.
New York, NY: John Wiley & Sons, Inc.
Carlson, Francis. 1974. Muscle Physiology. Englewood
Cliffs, New Jersey: Prentic-Hall, Inc.
Cooper, J.A. 2002. Actin Dynamics: Tropomyosin Provides Stability.
Current Biology 12, R523-R525.
Gordon, A.M; Regnier, M., Homsher, E. April 2001.Skeletal and
Cardiac Muscle Contractile Activation: Tropomyosin "Rocks and Rolls."
News in Physiological Sciences. Volume 16. 13 March 2003. <http://www.med.umich.edu/phys/578/articles/brooks%20apr10-1.pdf>
Kimball, John W. Muscles. 29 January 2003 John W. Kimball's
Biology Pages.13 March 2003. <http://biology.about.com/gi/dynamic/offsite.htm?site=http%3A%2F%2Fwww.ultranet.com%2F%7Ejkimball%2FBiologyPages%2FM%2FMuscles.html>
Kreis, Thomas; Vale, Ronald.1993. Guidebook to the
Cytoskeletal and Motor Proteins. New York, NY: Oxford University Press,
Inc.
Maciver, Sutherland. Lab Web Page. 2003. University of Edinburgh.
12 March 2003. <http://www.bms.ed.ac.uk/research/others/smaciver/Index.htm>
Maron, D. Hypertrophic Cardiomyopathy Association. 2002. 13
March 2003. < http://www.4hcm.org/overview/index.php>
Purves WK, David S, Orians GH, Heller HC. 2001. Life: The
Science of Biology 6th ed. Gordonsville, Virginia: Sinauer Associates,
Inc.
Stracher, Alfred. 1983. Muscle and Nonmuscle Motility.
New York, NY: Academic Press, Inc.
References for Images (in order of appearance)
Figure 1: Anonymous.Ohio State University Pages. 12 March
2003. <http://class.fst.ohio-state.edu/FST822/lectures/Muscl.htm>
Figure 2: Watkins, Hugh. Oxford University Cardiovascular
Research. 22 Sept. 2002 12 March 2003. <http://www.cardiov.ox.ac.uk/staff/hwg.html>
Figure 3: Barnay, M.; Barnay, K. Biochemistry of Muscle
Contraction. March 2002. The University of Illinois at Chicago. 12 March 2003.<http://www.uic.edu/classes/phyb/phyb516/regulatoryproteinsu.htm>
Figure 4: Anonymous. Ohio State University Lectures.13
March 2003.<http://class.fst.ohio-state.edu/FST822/lectures/Muscl.htm>
Figure 5: Heimberg, Thomas. 13 March 2003.<http://www.mpibpc.gwdg.de/inform/MpiNews/cientif/jahrg5/2.99/fig1.html>
Animation Link. Campbell, Malcolm. Molecular Biology
Web pages. Davidson College. 13 March 2003. <http://bio.davidson.edu/misc/movies/tropotropo.mov>
Figure 6: Rule, M. Biochemistry Undergraduate Course
Web page. 6 Jan. 2003. Carnegie Mellon University. 13 March 2003. <http://info.bio.cmu.edu/courses/03231/ProtStruc/2tma.htm>
Figure 7: Watkins, Hugh. Oxford University Cardiovascular
Research. 22 Sept. 2002 13 March 2003. <http://www.cardiov.ox.ac.uk/staff/hwg.html>

Davidson College Biology
Dept Web Page
Davidson Molecular
Biology Web Page
If you have any questions, suggestions, or comments, please
e-mail Maureen Keogh!