This web page was produced as an assignment for an undergraduate course at Davidson College.
Protein Crystallization
Background:
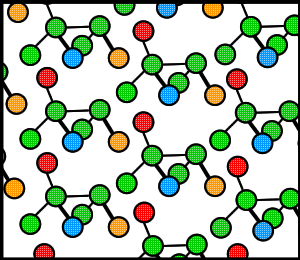
Proteins, like many molecules, can be prompted to form crystals when placed in the appropriate conditions. In order to crystallize a protein, the purified protein undergoes slow precipitation from an aqueous solution. As a result, individual protein molecules align themselves in a repeating series of "unit cells" by adopting a consistent orientation. The crystalline “lattice” (Fig. 1) that forms is held together by noncovalent interactions (Rhodes, 1993). The importance of protein crystallization is that it serves as the basis for X-ray crystallography, wherein a crystallized protein is used to determine the protein’s three-dimensional structure via X-ray diffraction. In 1934, Bernal and Hodgkin discovered that protein crystals surrounded by their mother liquor gave better diffraction patterns than dried crystals. Using pepsin (Fig. 2), they were the first to discern the diffraction pattern of a wet, globular protein. Prior to Bernal and Hodgkin, protein crystallography had only been performed in dry conditions with inconsistent and unreliable results (Tulinsky, 1999).
Figure 1. Cartoon of a hypothetical crystalline lattice. Colors have been added merely as reference points.
Initial progress was slow in discerning protein shapes, due to the difficulty inherent in crystallizing proteins. When the Protein Data Bank (http://www.pdb.org) was founded in 1971, it contained only seven structures (Berman, et al., 2000). Since then, the pace at which protein structures are being discovered has grown exponentially, with the PDB currently containing over 20,000 structures (PDB, 2003). Our ability to crystallize proteins has obviously improved, and this web page highlights a few of the methods currently available.
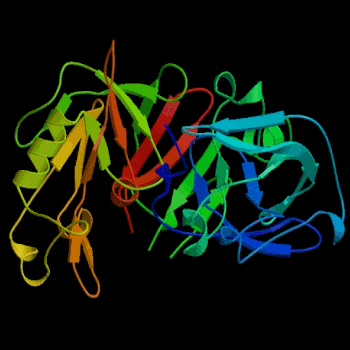
Figure 2. Three-dimensional structure of porcine pepsin. Pepsin was the first globular protein crystal used successfully in x-ray diffraction. Image courtesy of Protein Databank (ID: 3PSG; PDB, 2003).
Overview:
The goal of crystallization is usually to produce a well-ordered crystal that is lacking in contaminants and large enough to provide a diffraction pattern when hit with x-ray. This diffraction pattern can then be analyzed to discern the protein’s three-dimensional structure. Protein crystallization is inherently difficult because of the fragile nature of protein crystals. Proteins have irregularly shaped surfaces, which results in the formation of large channels within any protein crystal. Therefore, the noncovalent bonds that hold together the lattice must often be formed through several layers of solvent molecules (Rhodes, 1993). In addition to overcoming the inherent fragility of protein crystals, the successful production of x-ray worthy crystals is dependent upon a number of environmental factors because so much variation exists among proteins, with each individual requiring unique conditions for successful crystallization. Therefore, attempting to crystallize a protein without a proven protocol can be very tedious . Some factors that require consideration are protein purity, pH, concentration of protein, temperature, and precipitants. In order for sufficient homogeneity, the protein should usually be at least 97% pure. pH conditions are also very important, as different pH’s can result in different packing orientations. Buffers, such as Tris-HCl, are often necessary for the maintenance of a particular pH (Branden and Tooze, 1999). Precipitants, such as ammonium sulfate or polyethylene glycol, are compounds that cause the protein to precipitate out of solution (Rhodes, 1993).
Vapor Diffusion:
Two of the most commonly used methods for protein crystallization fall under the category of vapor diffusion. These are known as the hanging drop and sitting drop methods. Both entail a droplet containing purified protein, buffer, and precipitant being allowed to equilibrate with a larger reservoir containing similar buffers and precipitants in higher concentrations. Initially, the droplet of protein solution contains an insufficient concentration of precipitant for crystallization, but as water vaporizes from the drop and transfers to the reservoir, the precipitant concentration increases to a level optimal for crystallization. Since the system is in equilibrium, these optimum conditions are maintained until the crystallization is complete (Rhodes, 1993; McRee, 1993).
Simply put, the hanging drop method differs from the sitting drop method in the vertical orientation of the protein solution drop within the system. It is important to mention that both methods require a closed system, that is, the system must be sealed off from the outside using an airtight container or high-vacuum grease between glass surfaces. Figures 3 and 4 depict the hanging drop and sitting drop systems, respectively (Rhodes, 1993; McRee, 1993).
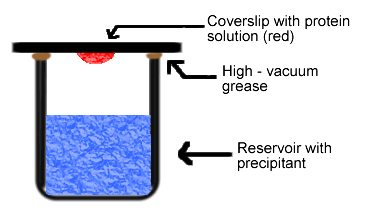
Figure 3. Diagram of hanging drop method. Reservoir solution (blue) usually contains buffer and precipitant. Protein solution (red) contains the same compounds, but in lower concentrations. The protein solution may also contain trace metals or ions necessary for precipitation of particular proteins. For instance, insulin is known to require trace amounts of zinc for crystallization (McRee, 1993).
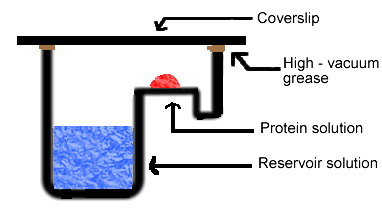
Figure 4. Diagram of sitting drop method. In this method, the protein drop sits on a pedestal above the reservoir solution, as opposed to hanging.
High Through-Put Methods:
One unavoidable aspect of crystallizing a protein from scratch is the need for a large number of experiments exploring the various conditions that are necessary for successful crystal growth. However, it is tedious to set up and screen so many different experimental conditions. Thus, high through-put methods exist to help streamline this process. There are numerous kits available to order which apply preassembled ingredients in systems guaranteed to produce successful crystallization. Using such a kit, a scientist avoids the hassle of purifying a protein and determining the appropriate crystallization conditions. Another method that has proven useful in setting up a vast number of crystallizations involves robotics. What would otherwise be slow and error-prone when carried out by a human can be accomplished efficiently and accurately with an automated system. Click here to see a movie of a crystallization robot in action (http://www.syrrx.com/technology/index.htm). Robotic crystallization systems use the same components described above, but carry out each step of the procedure quickly and with a large number of replicates. Each experiment utilizes tiny amounts of solution, and the advantage of the smaller size is two-fold: the smaller sample sizes not only cut-down on expenditure of purified protein, but smaller amounts of solution lead to quicker crystallizations. Each experiment is monitored by a camera which detects crystal growth (Eurekalert, 2000).
Links
A photo gallery of protein crystals.
NASA's protein crystal growth home page. (A microgravity environment is apparently conducive to successful crystal growth.)
Protein Crystallization and Dumb Luck. (An essay on the haphazard side of protein crystallization by Bob Cudney.)
**Unless otherwise noted, all figures created by John M. Kogoy.
References
Berman, Helen M., et al. "The Protein Data Bank." Nucleic Acids Research. 28: 235-242 (2000). <http://nar.oupjournals.org/cgi/content/abstract/28/1/235>
Branden, Carl and John Tooze. Introduction to Protein Structure. New York: Garland, 1999 (pp. 374-376).
"The Crystal Robot." Eurekalert. Accessed: February 18, 2003. <http://www.eurekalert.org/features/doe/2000-12/drnl-tcr061902.php>
McRee, Duncan E. Practical Protein Crystallography. San Diego: Academic Press, 1993 (pp. 1-23).
Protein Databank. Last updated: 2003. Accessed: February 17, 2003. <http://www.pdb.org>
Rhodes, Gale. Crystallography Made Crystal Clear. San Diego: Academic Press, 1993 (pp. 8-10, 29-38).
Tulinsky, A. "The Protein Structure Project, 1950-1959: First Concerted Effort Of a Protein Structure Determination In the U.S." The Rigaku Journal. 16 (1999). <http://www.rigakumsc.com/journal/Vol16.1.1999/tulinsky.pdf>