~ This web page was produced as an assignment for an undergraduate course at Davidson College ~
The Huntingtin gene and protein
A mutation in the IT15 gene (renamed huntingtin after discovering its role in Huntington's Disease) causes a mutation in the protein resulting from this gene, known as the huntingtin protein (abbreviation: Htt) thus causing Huntington's Disease. Huntington's disease is inherited as an autosomal dominant disease that gives rise to progressive, selective (localized) neural cell death leading to the classic signs of Huntington disease which are progressive chorea (a nervous disorder marked by spasmodic movements of limbs, facial muscles and incoordination), rigidity, and dementia, frequently associated with seizures. The age at onset is highly variable: some showed signs in the first decade and some not until over 60 years of age. The average is between 30 and 40 years old (Chandler et al., 1960). The clinical features develop progressively with severe increase in choreic movements and dementia and the disease terminates in death on average 17 years after manifestation of the first symptoms (Reed and Neel, 1959). The most noticeably affected functions are attention, learning, and planning.
In 1983, the Huntington's Disease gene was first mapped to the tip of the short arm of chromosome 4, however the gene was not completely isolated as the direct cause of Huntington's Disease until 1993. The Huntington's Disease Collaborative Research Group used haplotype analysis of linkage disequilibrium to focus in on a small segment of 4p16.3 as the likely location of the defect (MacDonald et al., 1992). The G8 locus and presumably the Huntington disease locus are deleted in the Wolf-Hirschhorn (4p-) syndrome (Gusella et al., 1985). This information helped map the HD locus to 4p, however most 4p- syndrome patients do not survive long enough to develop manifestations of HD. Another approach to locating the gene was done using a strategy of chromosome jumping to identify new probes from the terminal portion of 4p, which also confirmed the theory of location on the 4p16.3 segment (Collins et al., 1987).
The Huntington's Disease Collaborative Research Group (1993) found a 'new' gene designated IT15 (important transcript 15), later renamed huntingtin as mentioned above, which was isolated using cloned trapped exons from the target area. This gene contains a polymorphic trinucleotide repeat that is expanded and appeared unstable on Huntington Disease (HD) chromosomes. A CAG repeat - the nucleotide code for the glutamine amino acid - longer than the normal range was observed on HD chromosomes in exon 1, from all disease families examined. This repeat appeared to be located within the coding sequence of a predicted protein of about 348 kD that is widely expressed but unrelated to any known gene. Thus it turned out that the HD mutation involves an unstable DNA segment similar to those that can be found in several other disorders, such as the fragile X syndrome, Kennedy syndrome, and myotonic dystrophy. The fact that the phenotype of HD is completely dominant suggests that the disorder results from a gain-of-function mutation in which either the mRNA product or the protein product of the disease allele has some new property or is expressed inappropriately (Myers et al., 1989).
The repeat is mutated during meiosis, with a higher mutation rate in genes that already have a high number of trinucleotide repeats, such that those who already have the requisite number of repeats to cause HD are more likely to have mutations that expand the repeated sequence even more. The risk of the expansion mutations were also found to be more frequent during spermatogenesis compared to oogenesis, perhaps because many more sperm are made than eggs, allowing more room for mistakes. The gene was proven to be mitotically stable, and the number of repeated triplets was found to be consistent among various types of cells within the body. Read (1993) summarized and collated results from 3 different studies showing that the normal range of repeat numbers was 9-11 at the low and 34-37 at the high end, with a mean ranging from 18.29 to 19.71. Duyao et al. (1993) found a range of 37-86 in HD patients with a mean of 46.42. The HD mutation is not fully penetrant in individuals with a borderline number of CAG repeats, thus those individuals who have approximately 36-39 repeats may show some symptoms of HD while never having a complete onset of the disease. The extent of expansion is correlated with both age of onset and severity of disease such that juvenile onset HD patients have extremely large CAG expansions (up to 100) and a very severe form of the disease. The DNA sequence of the entire gene is a major influence on the CAG stability, especially in relation to several adjacent CCG tracts which affected the rate of expansion in the trinucleotide repeat.
Ambrose et al. (1994) found that the HD locus spans 180 kb and contains 67 exons ranging in size from 48 bp to 341 bp with the average size of 138 bp. The gene is ubiquitously expressed as 2 alternatively polyadenylated forms showing different relative abundances in various fetal and adult tissues. The function of the normal gene is not yet known, except that the protein expressed from the gene can be found in large quantities around the body and has been associated with transcriptional repression. Several recent studies have also found a link to dopamine receptor expression as noted in Fig. 3 below.
At the bottom of this link to an NCBI page you will find the gene sequence for huntingtin. The huntingtin gene sequence. Also of interest halfway down the page is the amino acid sequence for the huntingtin protein in which the glutamine repeat can be noted on the first line as a repetition of 'Q's, the one-letter amino code for glutamine (CAG).
Western blot experiments were performed with monoclonal antibodies using the known sequence from the huntingtin gene detecting the huntingtin protein at approximately 350 kD in various human cell lines and in neural and non-neural rodent tissues (for sequence of protein see link to NCBI). In cell lines from HD patients, a doublet protein was detected corresponding to the mutant and normal huntingtin, with the larger band showing the relative increase in molecular weight proportionally due to the number of CAG repeats. Polyclonal and monoclonal antibodies have been used to show native huntingtin orthologs in rat as well as monkey. The huntingtin protein is located in neurons throughout the brain, with the highest levels evident in the larger striatal neurons. In humans, huntingtin was revealed in a patch-like distribution, potentially corresponding to the first areas affected in HD. The protein appears to be associated particularly with microtubules, although some is also associated with synaptic vesicles and based on this observation of association, it has been suggested that the mutation affects cytoskeletal anchoring. Mutant Htt concentrates in the nucleus of affected neurons, whereas normal Htt congregates mainly in the cytoplasm usually on one face of the nucleus, however the normal Htt protein can be found in very small amounts in the nucleus as well (Tao and Tartakoff, 2001).

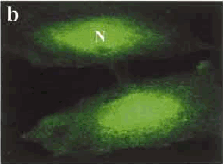
Fig. 2: (a) Staining of a cell producing normal Htt protein, with most being found in the cytoplasm, on one side of the nucleus (N) wall. (b) Staining of a cell with mutated DNA causing accumulation of the Htt protein inside the nucleus. (Images taken from Tao and Tartakoff, 2001)
The glutamine residues encoded by CAG repeats are involved in the formation of cross-links within and between proteins, through a reaction catalyzed by transglutaminases (TGases). Cariello et al. (1996) speculated that TGase may be involved in the molecular process of neurodegeneration in HD since longer polyglutamine stretches may be better substrates for TGases; increased glutamine cross-linking could induce the formation of rigid supramolecular structures, with consequent neuronal death. When noting the aggregation of proteins in the nucleus, it was found that the rate of aggregation was dependent on the number of repeats, and the presence of wildtype huntingtin neither enhanced nor interfered with protein aggregation.
Work done by Zuccato et al (2001) demonstrated that wildtype huntingtin upregulates transcription of brain-derived neurotrophic factor (BDNF), a prosurvival factor produced by cortical neurons that is necessary for survival of striatal neurons in the brain. When the huntingtin protein is mutated, production of this factor is decreased which leads to insufficient support for the striatal neurons, causing their death.
Full-length wildtype and mutant huntingtin repressed transcription when targeted to DNA, but truncated N-terminal wildtype huntingtin did not, suggesting that proteolysis (protein degradation) of huntingtin in the nucleus may normally occur in cells to terminate or modulate huntingtin function. However, truncated N-terminal mutant huntingtin retained the ability to repress transcription, suggesting an abnormal gain-of-function (Kegel et al., 2002).
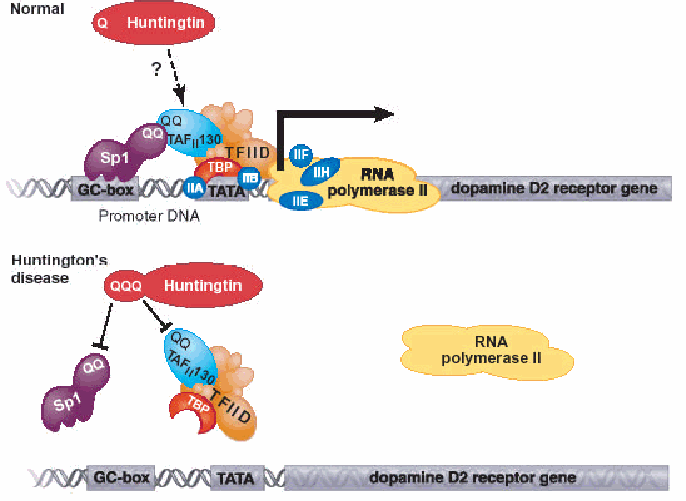
Fig. 3: Top: The transcriptional factor Sp1 binds to DNA elements called GC boxes in cellular promoters. A specific protein-protein interaction between the glutamine-rich (QQ) regions in Sp1 and the TAFII 130 subunit is required for recruitment of the general transcriptional machinery. The glutamine interface serves to bridge Sp1 to the machinery required to recruit RNA polymerase II in order for transcription into mRNA to begin. When the Huntingtin protein is mutated, the RNA polymerase II cannot bind properly thus transcription of the dopamine receptor gene does not occur. (Permission for image pending from a Cornell lecture)
The genetic defect leading to Huntington's disease may not necessarily always eliminate transcription, but may confer a new property on the mRNA or alter the function of the protein. One candidate is the huntingtin-associated protein-1, highly expressed in brain, which has increased affinity for huntingtin protein with expanded polyglutamine repeats. Another possibility is that instead of looking in the nucleus or cytosol, some authors suggest that polyglutamine stretches of about 40 residues or more can span the lipid bilayer in a helical form to make an ion channel. The most interesting feature of the helix is that it does not form several transmembrane helices, but rather one large, tubular pore, which could allow positive molecules and water molecules across the membrane and harm the ion gradients in the cell. However this latter proposal is not as well supported as the investigations into protein aggregation. Htt also often interacts with a protein known as GAPDH - a key enzyme in glycolysis, and mutated huntingtin protein disrupts the cAMP response element binding protein which along with disrupting BDNF activities alters neurotransmission and cellular metabolism. Other proposed mechanisms contributing to the neural degeneration include excitotoxicity, oxidative stress, and apoptosis (Ho et al, 2001).
Links of Interest:
HotMolecBase review for Huntingtin
Huntington's Disease Society of America
The Huntington's Disease Association Online (UK)
Huntington's Disease Advocacy Center
- Ambrose, C. M.; Duyao, M. P.; Barnes, G.; Bates, G. P.; Lin, C. S.; Srinidhi, J.; Baxendale, S.; Hummerich, H.; Lehrach, H.; Altherr, M.; Wasmuth, J.; Buckler, A.; Church, D.; Housman, D.; Berks, M.; Micklem, G.; Durbin, R.; Dodge, A.; Read, A.; Gusella, J.; MacDonald, M. E. "Structure and expression of the Huntington's disease gene: evidence against simple inactivation due to an expanded CAG repeat". Somatic Cell & Molecular Genetics; 1994, 20: 27-38.
- Cariello, L.; de Cristofaro, T.; Zanetti, L.; Cuomo, T.; Di Maio, L.; Campanella, G.; Rinaldi, S.; Zanetti, P.; Di Lauro, R.; Varrone, S. "Transglutaminase activity is related to CAG repeat length in patients with Huntington's disease". Human Genetics; 1996, 98: 633-635.
- Chandler, J. H.; Reed, T. E.; Dejong, R. N."Huntington's chorea in Michigan". Neurology; 1960, 10: 148-153.
- Collins, F. S.; Richards, J. E.; Cole, J. L.; Gilliam, T. C.; Gusella, J. F. "Chromosome jumping from D4S10 (G8) toward the Huntington disease gene". Cytogenet. Cell Genet; 1987, 46: 597.
- Duyao, M. P.; Auerbach, A. B.; Ryan, A.; Persichetti, F.; Barnes, G. T.; McNeil, S. M.; Ge, P.; Vonsattel, J.-P.; Gusella, J. F.; Joyner, A. L.; MacDonald, M. E. "Inactivation of the mouse Huntington's disease gene homolog Hdh". Science; 1995, 269: 407-410.
- Gusella, J.; Tanzi, R. E.; Bader, P. I.; Phelan, M. C.; Stevenson, R.; Hayden, M. R.; Hofman, K. J.; Faryniarz, A. G.; Gibbons, K. "Deletion of Huntington's disease linked G8 (D4S10) locus in Wolf-Hirschhorn syndrome". Nature; 1985, 318: 75-78.
- Ho, L. W.; Carmichael, J; Swartz J; Wyttenbach A; Rankin J; Rubinsztein DC. "The molecular biology of Huntington's disease." Psychologocial Medicine; 2001, 31(1): 3-14.
- Kegel, K. B.; Meloni, A. R.; Yi, Y.; Kim, Y. J.; Doyle, E.; Cuiffo, B. G.; Sapp, E.; Wang, Y.; Qin, Z.-H.; Chen, J. D.; Nevins, J. R.; Aronin, N.; DiFiglia, M. "Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription". Journal of Biological Chemistry; 2002, 277: 7466-7476.
- Myers, R. H.; Leavitt, J.; Farrer, L. A.; Jagadeesh, J.; McFarlane, H.; Mastromauro, C. A.; Mark, R. J.; Gusella, J. F. "Homozygote for Huntington disease". The American Journal of Human Genetics; 1989, 45: 615-618.
-MacDonald, M. E.; Novelletto, A.; Lin, C.; Tagle, D.; Barnes, G.; Bates, G.; Taylor, S.; Allitto, B.; Altherr, M.; Myers, R.; Lehrach, H.; Collins, F. S.; Wasmuth, J. J.; Frontali, M.; Gusella, J. F. "The Huntington's disease candidate region exhibits many different haplotypes". Nature Genetics; 1992, 1: 99-103.
- Read, A. P. "Huntington's disease: testing the test". Nature Genetics; 1993, 4: 329-330.
- Reed, T. E.; Neel, J. V. "Huntington's chorea in Michigan. II. Selection and mutation". The American Journal of Human Genetics; 1959, 11: 107-136.
- Tao, Tao; Tartakoff, A. M.. "Nuclear Relocation of Normal Huntingtin". Traffic; 2001, 2: 385-394.
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B. R.; Goffredo, D.; Conti, L.; MacDonald, M. E.; Friedlander, R. M.; Silani, V.; Hayden, M. R.; Timmusk, T.; Sipione, S.; Cattaneo, E. "Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease". Science; 2001, 293: 493-498.
![]()
![]()
Comments, questions, and suggestions to: Megan McDonald