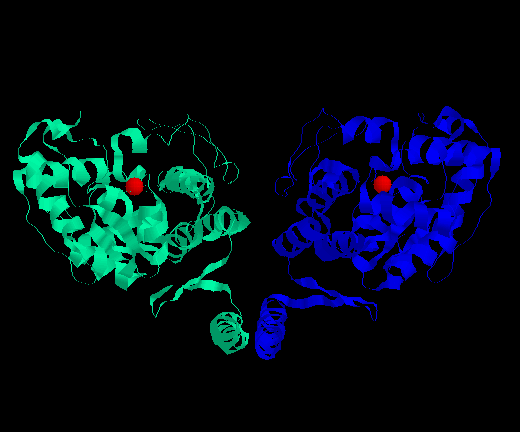
This page was created as an undergraduate assignment for a molecular biology course.
Presented by Isaac Miller
Figure 1: A look at two of the four monomers making up a functional tetrameric PAH unit. Iron III ions present in the active sites are shown in red. Image produced using RasMol and structural data in PDB format from www.rcsb.org/pdb.
Introduction:
Phenylalanine hydroxylase (PAH) is a protein that is highly conserved among animals; this conservation in mammals can be seen in the alignment of human, rat, and mouse PAH sequences in the figure shown on Phenylalanine Hydroxylase Locus Knowledgebase at http://www.pahdb.mcgill.ca/pahdb_alignment.html. PAH is also found in other kingdoms, such as bacteria, as is shown in Figure 2, though the sequences between PAH in proteins and animals are significantly different. The protein functions primarily to convert the amino acid phenylalanine into tyrosine with the addition of oxygen and a pterin cofactor, as shown in Scheme 1. Mutations in the gene that codes for PAH lead to a condition known as Phenylketonuria (PKU), which can cause mental retardation and other effects in those suffering from the disease.
Figure 2. Amino acid sequence alignment of PAH in Chromobacterium violaceum, rats, and humans. Human and rat PAH sequence is highly conserved, while the bacterial sequence is different, but is significantly (24%) homologous to the rat sequence similarly homologous to the human sequence. Boxes indicate sequence conserved in all three species, periods are spacers to maximize overlap, and asterisks indicate residues identical to TrpOHase and TyrOHase, similar hydroxylating enzymes that work on different amino acids (Figure taken from Onishi et al. 1991).
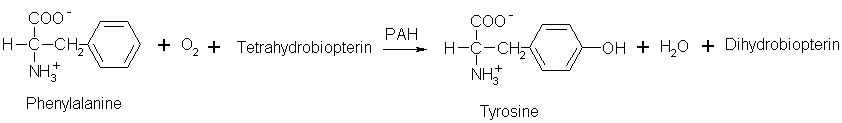
Scheme 1. PAH acts with oxygen and tetrahydrobiopterin cofactor to convert phenylalanine into tyrosine. Scheme produced with IsisDraw 2.4; information on reaction from http://us.expasy.org/cgi-bin/nicezyme.pl?1.14.16.1.
PAH exists in activated and inactivated forms, and can be activated by different types of molecules including detergents and amino acids. Activation by phenylalanine itself is the only activating pathway that has been seen in living cells and is presumably the natural activation pathway. A phenylalanine molecule binds to an activation site but is not hydroxylated. The activated enzyme can then hydroxylate other phenylalanine molecules as long as it remains activated by the phenylalanine ligand (Shiman, et al. 1990). This is only one aspect of a complicated mechanism that involves cofactors and activation ligands and is not well understood.
Mutations have been documented at many points on the human PAH gene. For the full-length human gene, see the Phenylalanine Hydroxylase Locus Knowledgebase genome page, and for a map of reported mutations, see their mutation map (2000).
Mutations that render the enzyme nonfunctional or significantly reduce its efficiency to below 20% cause Phenylketonuria (Johns Hopkins University OMIM 2003), or PKU, a recessive disorder which requires the inheritance of two damaged alleles and strikes approximately 1 of every 10,000 people of European descent (Nowacki 1998).
In a person with PKU, phenylalanine builds up in the bloodstream, which, by unknown mechanism, inhibits normal brain development and results in severe mental retardation unless a special low-phenylalanine diet is started at birth. Lack of tyrosine is not an issue, even though phenylalanine cannot be converted to tyrosine, since enough is obtained in a normal diet (Nowacki 1998).
Treatment of PKU consists of a low-phenylalanine diet, which is particularly important during early childhood when the brain is developing rapidly. Much debate concerns whether an extremely restrictive low-phenylalanine diet should be maintained throughout a patient's life, but current data suggests that maintaining the diet leads to increased ability to concentrate and other benefits for older sufferers (Nowacki 1998).
A particular concern for women suffering from PKU is that their children, even if they do not have PKU themselves, will suffer impaired brain development due to the high levels of phenylalanine in the mother's blood. This is compounded by the fact that higher levels of amino acids are maintained in the fetus than in the mother's own blood. Thus, women who suffer from PKU and wish to have children should be particularly strict in maintaining a low-phenylalanine diet even before conception to insure healthy development of her child (Nowacki 1998).
References:
Debelle Databases Server. 2000. PAHdb: Mutation Map. Phenylalanine Hydroxylase Locus Knowledgebase. <http://www.pahdb.mcgill.ca/pahdb_mutationmap.html> Accessed 2003 Mar 13.
Debelle Databases Server. 2000. Sequence. PAHdb: Phenylalanine Hydroxylase Locus Knowledgebase. <http://www.pahdb.mcgill.ca/pahdb_sequence.html> Accessed 2003 Mar 13.
ExPASy Molecular Biology Server. 2003. NiceZyme View of ENZYME: EC 1.14.16.1. <http://us.expasy.org/cgi-bin/nicezyme.pl?1.14.16.1> Accessed 2003 Mar 13.
Johns Hopkins University. 2003. Phenylketonuria. OMIM: Online Mendelian Inheritance in Man. <http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=261600> Accessed 2003 Mar 13.
Nowacki PM. 1998. Montreal Children's Hospital: Hyperphenylalaninemia (PKU) Resource Booklet for Families. <http://www.pahdb.mcgill.ca/handout/handout.htm#What%20is%20PKU> Accessed 2003 Mar 13.
Onishi A, Liotta LJ, Benkovic SJ. 1991. Cloning and Expression of Chromobacterium violaceum Phenylalanine Hydroxylase in Escherichia coli and Comparison of Amino Acid Sequence with Mammalian Aromatic Amino Acid Hydroxylases. J. Biol. Chem. 266(28):18454-18459.
Shiman R, Jones SH, Gray, DW. 1990. Mechanism of Phenylalanine Regulation of Phenylalanine Hydroxylase. J. Biol. Chem. 265(20):11633-11642.
Return to my Web Page List
Send questions or comments to Isaac Miller
Last modified: Mar. 13, 2003
