This page was produced as an assignment for an undergraduate course at Davidson College
WHAT ARE PRIONS?
Prions are small, simple proteins. They are infective, and interesting because they are the only known infective agents that do not contain genetic material. Prions are transmissible particles that are devoid of nucleic acid and seem to be composed exclusively of modified protein PrPSc (Prusiner, 2003). Prion is an acronym for, proteinaceous infective particle.
PAST PRIONS
Prions are important because when they change conformation, they cause problems. Normal versions of cellular prion proteins are abundant on the surface of brain nerve cells and are said to be involved in synaptic function. These wild type prion proteins, PrPC, were discovered upon investigation of the diseased version that causes neurodegenerative diseases, PrPSc. Transmissible spongiform encephalopathies (TSE's) are degenerative brain diseases that occur in many mammals. The brain degrades gradually, developing holes as it does so and evetually looks like a sponge. The three most notably afflicted animal species affected by the disease causing form of prion proteins, PrPSc , are Ovis aries (sheep), Bos taurus (cows) and Homo sapiens. Prion proteins exist as 804 bp DNA in each of the aforementioned (NCBI) but the protein structures differ slightly. The protein structure of prions in sheep involves 256 amino acids, 253 in humans and 264 in bovine. The orthologs are as follows:
1 mvkshigswi lvlfvamwsd vglckkrpkp gggwntggsr ypgqgspggn ryppqggggw
61 gqphgggwgq phgggwgqph gggwgqphgg gwgqphgggg wgqggthgqw nkpskpktnm
121 khvagaaaag avvgglggym lgsamsrpli hfgsdyedry yrenmhrypn qvyyrpvdqy
181 snqnnfvhdc vnitvkehtv ttttkgenft etdikmmerv veqmcitqyq resqayyqrg
241 asvilfsspp villisflif livg
1 mvkshigswi lvlfvamwsd vglckkrpkp gggwntggsr ypgqgspggn ryppqggggw
61 gqphgggwgq phgggwgqph gggwgqphgg ggwgqggshs qwnkpskpkt nmkhvagaaa
121 agavvgglgg ymlgsamsrp lihfgndyed ryyrenmyry pnqvyyrpvd qysnqnnfvh
181 dcvnitvkqh tvttttkgen ftetdikime rvveqmcitq yqresqayyq rgasvilfss
241 ppvillisfl iflivg
1 manlgcwmlv lfvatwsdlg lckkrpkpgg wntggsrypg qgspggnryp pqggggwgqp
61 hgggwgqphg ggwgqphggg wgqphgggwg qgggthsqwn kpskpktnmk hmagaaaaga
121 vvgglggyml gsamsrpiih fgsdyedryy renmhrypnq vyyrpmdeys nqnnfvhdcv
181 nitikqhtvt tttkgenfte tdvkmmervv eqmcitqyer esqayyqrgs smvlfssppv
241 illisflifl ivg
Scrapie is the TSE that is caused by PrPSc proteins in sheep and goats. It has been observed for 250 years (Purves, 2001). In the 1980s, Brittish cows started to display some of the same bazzar behavior that PrPSc prone sheep had previously. The TSE in cows was traced to ingestion of sheep products. Ten years later, humans displayed TSEs. This "Mad Cow disease" was traced to ingestion, by humans, of diseased beef and suggested the crossing of species lines.
Kuru, one of five forms of human TSEs had previously been seen in the 1950s where tribal ritual required that brains of the deceased not be burried, but used as food. Kuru was indigenous to New Ginea. This was unfortunate, but it led to realization and research establishing TSEs' ability to be transmitted from one animal to another via brain extracts from a diseased animal. Along with kuru, prions cause Alpers Syndrome, GSS - Gerstmann-Straussler-Scheinkner Syndrome, FFI - Fatal Familial Insomnia and CJD - Creutzfeld-Jacob Disease (Prusiner, 2003).
Transmitted extracts of brain tissue continued to cause TSEs even after being exposed to high doses of ultraviolet light to inactivate nucleic acids. This suggested that the causitive agent was not viural which would have contained denatured nucleic acid. Furthermore, because the amino acid sequence of proteins found in healthy brain tissue was the same as that of the proteins found in TSE brain tissue, TSE was deemed not to be caused by a mutated gene. Primary structure is the same, but the tertiary structures of PrPSc and PrPC are different and has a tremendous effects on the protein's function in the cell. The abnormal protein appears to induce conformational change in the normal PrPC protein so that it also becomes abnormal (Purves, 2001).
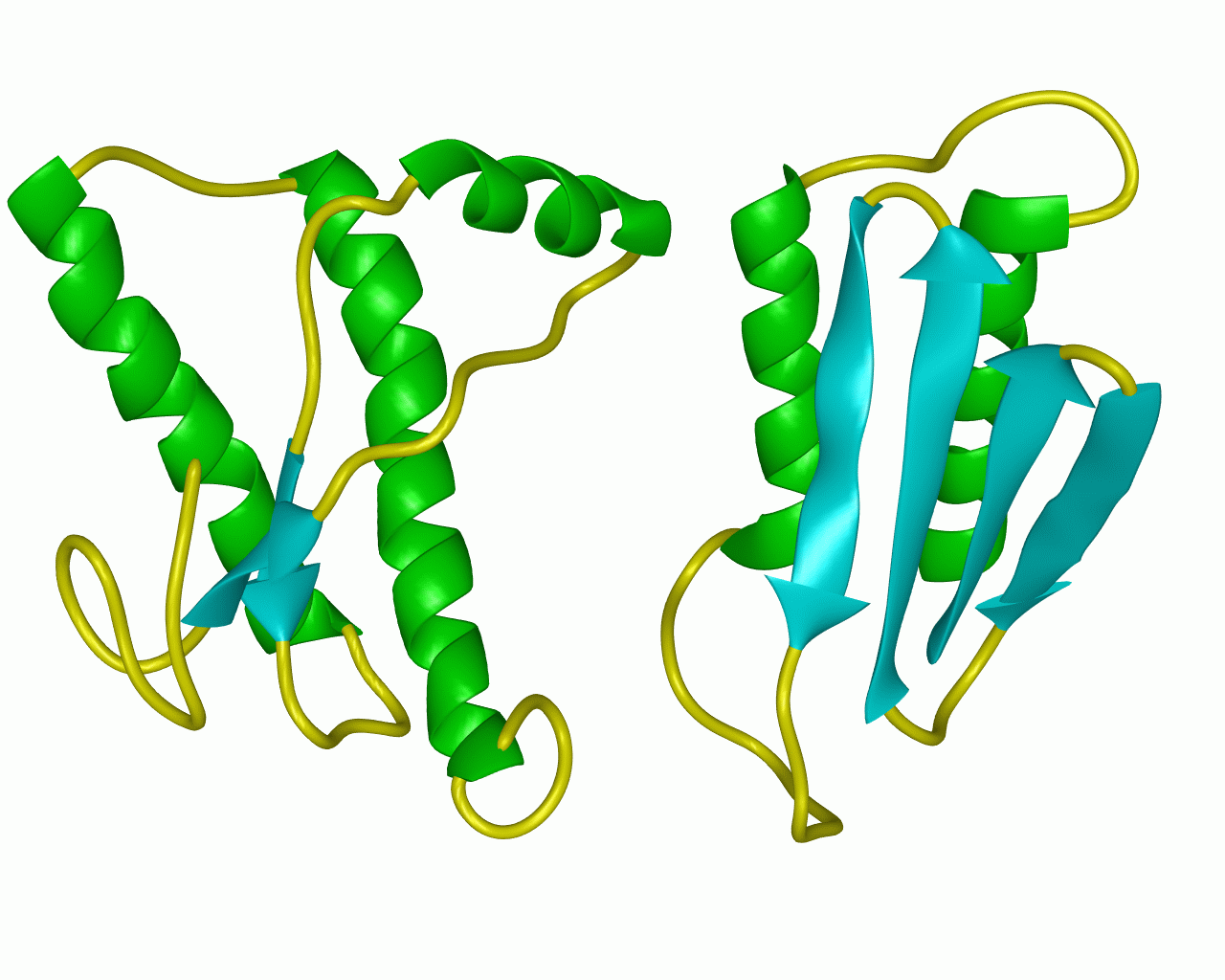
figure 2: the normal prion protein (left) has many alph-helix regions and is fairly soluble while the abnormal prion protein (right) has many beta-pleated sheet regions and is insoluble
WHAT'S NEW
Stanley B. Prusiner (see photo) was awarded a Nobel Prize in 1997 for his discovery of prions. In a recent article (see article) Prusiner established the relationship between two of the nervous system degenerative disorders caused by prions: Bovine spongiform encephalopathy (BSE) and (CJD).
In March 1996, the same year a ban was put on Brittish beef, ten people were identified with a new form of CJD. It differed from the known, sporadic form of the disease in both clinical and neuropathological features and was called new variant CJD (vCJD). vCJD and BSE are thought to be caused by the same strain of agent. There are at least three prion protein subtypes distinguished by two sites for sugar attachment. Diglycosolated, monoglycosolated and unglycosolated sugars are found in similar ratios in vCJD and BSE though both differ more dramatically from the classical forms of CJD. Patterns of neuropathological lesion profiles were determined using infection material and mice and patterns with vCJD and BSE were similar to eachother, but different from sporadic CJD.
brain afflicted by an infective protein prion:
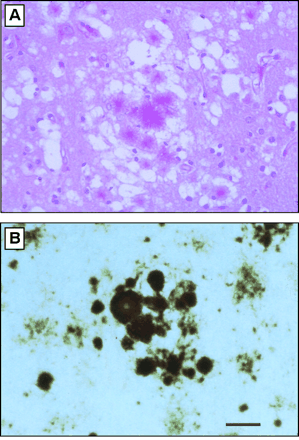
figure 3: Histopathology of vCJD in Great Britain. (A) Section from frontal cortex stained by the periodic acid-Schiff (PAS) method, showing a field with aggregates of plaques surrounded by spongiform degeneration. (B) Multiple plaques and amorphous deposits are PrP-immunopositive. Scale bar, 50 µm. Photomicrographs prepared by S. J. DeArmond. (Prusiner, 2003)
There is hope. Many methods of research are being employed for investigation, including: PrP gene analysis, PrP immunocytochemistry and detection by Western blot, histoblot, and immunoblot techniques, prion rod/SAF detection by electronmicroscopy, and transmissibility to both wild-type and transgenic laboratory animals all now have diagnostic applications (Baker, 1996).
Prions break rules. They are deviod of nucleic acid. Sheep and bovine prions have similar amino acid sequences, but it is the TSEs of bovine and humans that appear to be most closely correlated. If the PrPSc protein is the only infectious protein that causes disease in vertebrates (Hall, 2002), what does the normal wild-type version do? We all have the prion protein in us, but no one knows exactly what its usual function is. In very rare instances it undergoes a change in conformation and then causes a disease. Currently, it is believed to be involved in copper regulation. Metal binding to a protein backbone is sensitive to acidic changes and prion protein may be part of a system to transport copper in and out of cells by varrying acidity (Hall, 2002). It is possible that glutamine, part of a repeat sequence in all prion proteins, but not involved in copper binding, is responsible for misfolded PrPSc prion proteins. Glutamine could also be the cause of conformational transitions thought to be the prime mechanism of prion diseases, and the protein-protein interactions that are believed to spread TSEs.
References:
Baker, Harry F, Ridley, Rosalind M, ed. Prion Diseases.1996. Totowa, New Jersey: Humana Press.
Becker, Owen M, Levy, Yaakov, 2002. Confrontational Polymorphism of Wild-Type and Mutant Prion Proteins: Energy Landscape Analysis. Proteins: Structure, Funtction, and Genetics.<http://physics.ucsd.edu/~klevy/prion_proteins.pdf>. Accessed 2003 Mar 12.
Hill, Linley E, 2002. Researchers Find Clues to the Normal Function of Prion Proteins <http://www.ucsc.edu/currents/01-02/06-03/prion.html> Accessed 2003 Mar 12.
Link Between BSE andCJD.<http://www.portfolio.mvm.ed.ac.uk/studentwebs/session2/group4/link.htm> Accessed 2003 Mar 12.
NCBI. <http://www.ncbi.nlm.nih.gov> Accessed Mar 10
OMIM. <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM> Accessed Mar 10.
Prusiner, Stanley B, 2003. Prion Diseases and the BSE Crisis. Science. <http://www.sciencemag.org/feature/data/prusiner/245.shl> Accessed 2003 Mar 12.
Purves WK, David S, Orians GH, Heller HC Life: The Science of Biology 6th ed. 2001. Gordonsville, Virginia: Sinauer Associates, Inc. p. 334.
Rhodes, Richard, 1997. Deadly Feasts: Tracking the Secrets of a Terrifying New Plague. 1997. New York: Simon & Schuster.
Davidson College Molecular Biology Homepage
Send Questions/Comments to Emily Wilson