Overview:
Human factor VIII, also called anti-hemophilic
factor, is a plasma glycoprotein that performs a substantial role in blood-clotting
(Pratt, 1999). By being found in the blood plasma,
factor VIII is able to get to any part of the body that may be bleeding.
Without being localized in the blood, this factor would have to be expressed
in every cell of the body to ensure bleeding could be stopped anywhere it
started. Human factor VIII's amino acid sequence is 216aa long. This amino
acid sequence is derived from an mRNA sequence containing 2536bp (NCBI,
2003). However, the coding sequence for the protein is only 651bp long.
This protein is not only found in humans though. Many other creatures, dogs
and mice
included, also use a form of factor VIII in blood-clotting. In these other
organisms the amino acid sequence is conserved, but the cDNA and mRNA varies
slightly.
Blood
Coagulation in mammals:
Blood clotting in mammals results
from the conversion of a soluble plasma protein, into an insoluble matrix
of fibers (Fig. 1). Fibrinogen or factor I is the soluble plasma protein
that is eventually converted into fibrin, the insoluble matrix (Ratnoff,
1978). The change of fibrinogen into fibrin requires many other components
that all play a role in the change.One protein that acts on the fibrinogen
is thrombin.
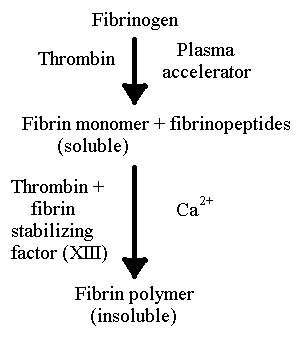
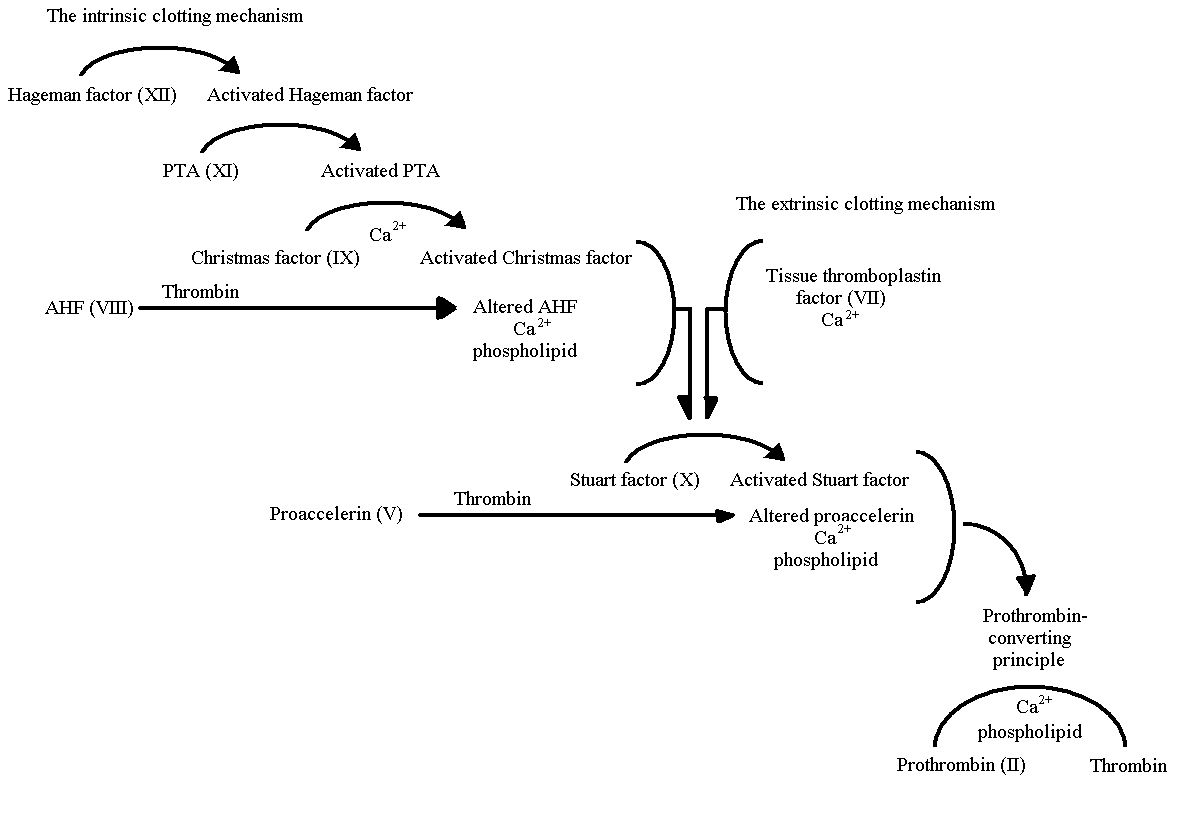
Fig. 2. The mechanisms of normal coagulation. Factor VIII
shown interacting to help form thrombin(Ratnoff,
1978).
In order for factor VIII to form a functional
protein, it must be activated by Christmas factor on the surface of phospholipids,
along with calcium ions being present. However, Factor VIII circulates through
the blood not as a solitary unit, but as a complex paired with von Willebrand
factor (Pratt, 1999). As shown in Fig. 2., after
factor VII is cleaved by thrombin, it binds with factor IX at either the
surface of activated platelets or endothelial cells. Factor IX is then able
to activate factor X, which in turn converts prothrombin to thrombin in
the presence of factor V. With calcium present, the waterfall activation
sequence is able to continue, however, without it, activated factor VIII
is not produced. Without the activation of factor VIII, prothrombin is never
changed into thrombin and clotting will not function properly (Pratt, 1999).
Disorders related
to Factor VII:
The main disorder that is related to factor
VII is Hemophilia A. Hemophilia A is a blood disorder that affects many
people through-out the world. In the United States alone this disorder affects
more than 20,000 people, along with about 400 babies being born with the
disorder (MSN Encarta, 2003). Hemophilia A is known
to cause, among other things, hemorrhaging into joints and muscles, easy
bruising, and prolonged bleeding from wounds. Hemophilia A is a X-linked
recessive disorder caused by a deficiency in the blood-clotting protein
coagulation factor VIII (OMIM, 2003). This disorder
has been traced to heterogeneous mutations in the factor VIII gene, located
at Xq28(OMIM, 2003). The severity of the disorder has been shown to be inversely
related to the "amount of residual factor VIII (<1%, severe; 2-5%,
moderate; and 5-30%, mild)" (OMIM, 2003). Hemophilia can also lead
to further ailments, including swelling, pain, decreased function, and degenerative
arthritis in the joints of the ankles, hips and knees (OMIM, 2003). Muscle
hemorrhage, another secondary ailment due to Hemophilia A, can also cause
necrosis, contractures, and neuropathy (OMIM, 2003).
The origin of Hemophilia A may be in the original
binding of factor VIII to von Willebrand factor. In order for prothrombin
to be changed into thrombin, the entire cascade of activations is dependent
on factor VIII binding to the von Willebrand factor. This binding has been
shown to be dependent upon the C2 region of factor VIII, shown in figure
3 (Pratt, 1999). Structurally destabilizing mutations in this area of the
the protein could cause factor VIII to become unable to bind with von Willebrand
factor and lead to the bleeding disorders of Hemophilia A (Pratt, 1999).
To view an in-depth video on Hemophilia click
here
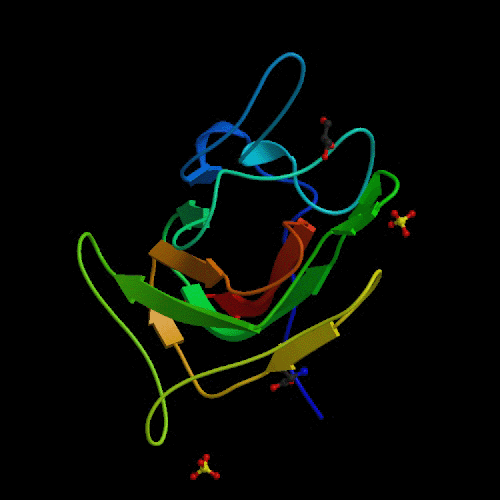
Fig.
3. Ribbon diagram of the C2 domain of Factor VIII.
Resources
MSN Encarta. 2003. Hemophilia. <http://www.nhlbi.nih.gov/health/public/blood/other/hemophel.htm>.
Accessed 2003 Mar 11.
NCBI. 2003. Hemophilia A. <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=nucleotide&list_uids=18490689&dopt=GenBank>.
Accessed 2003 Mar 11.
OMIM. 2003. Hemophilia A. <http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=306700>.
Accessed 2003 Mar 11.
Pratt, K.P., Shen, B.W., Takeshima, K., Davie,
E.W., Fujikawa, K., and Stoddard, B.L. 1999. Structure of the C2 domain
of human factor VIII at 1.5 Å resolution. Nature. 402:439-441.
Ratnoff, O.D., and Bennett, B., 1973. The Genetics
of Hereditary Disorders of Blood Coagulation. Science. 179:1291-1298.