*This web page was created as an assignment for an undergraduate course at Davidson College*
Human Hemochromatosis Protein
Introduction
Iron is an element that is crucial in the normal function of the body. Its biggest role is in oxidation-reduction reactions. Generally, the body absorbs dietary iron through the duodenum (a section of the intestine), and stores it in the heart, liver, and pancreas. Many different proteins are responsible for iron uptake and storage and some uncertainty exists as to the exact role all of them play in the process. One protein that is known to play a role in regulation iron concentration in cells is the Human Hemochromatosis protein, or HFE protein. When mutated, this protein causes build up of iron and eventual organ damage in the liver, heart, and pancreas. (Davies and Enns, 2004). Consequently, understanding the structure and function of this protein is important for human health.
Structure
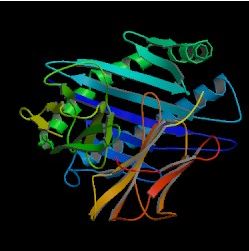
Image from the Protein Data Bank.
Figure 1. The basic structure of the HFE protein.
MHC class I Homologue
The HFE protein has a structure very similar to the major histocompatibility complex (MHC) class I molecules (The Hemochromatosis Gene) . Two alpha (alpha1 and alpha2) extracellular domains sit on top of a third alpha (alpha3) domain, which spans the cellular membrane and binds to a beta-2-microglobulin (beta2m) protein (The Hemochromatosis Gene). The first two alpha domains consist of two alpha helices on top of eight beta pleated sheets. The helices and sheets are anti parallel. An important disulfide bond occurs between C260 and C203 in alpha3; in the alpha1, H41 and A73 form a salt bridge (Lebron et al., 1998). Both of these interactions contribute to the overall structure of the protein.
Histidine Cluster
A patch of four histidine residues with a nearby tyrosine (Y118) is present in alpha1. This conformation is notable because it is similar to some iron-binding sites in other proteins. Although data have yet to indicate such a role at this site, repeat experiments at different pH's may indicate that the histidine cluster and Y118 do indeed bind iron. If so, this grouping could be a pH-dependent switch for the activity of the HFE protein (Lebron et al., 1998).
Lacks a Peptide Binding Grove
Most class I MHC molecules have a peptide binding grove, but because the alpha1 and alpha2 helices are closer in HFE, the analogous site in this protein is too narrow for peptide binding. This narrowing burries many residues that would otherwise participate in peptide binding and also causes steric clashes that prevent binding (Lebron et al., 1998).
Function
Generally, MHC class I proteins are responsible for bringing antigens to T-cells (The Hemochromatosis Gene). The function of the HFE protein is still under investigation, but despite its homology to these proteins, it appears to have no immune system function. Instead, it regulates iron concentration through different mechanisms in different cell types. In some cells it decreases iron concentration while in others it increases it (Davies and Enns, 2004).
Interaction with TfR and Tf
Iron (Fe) enters cells when diferric transferrin (Tf) binds to transferrin receptors (TfR), which brings the iron into the cell (Niemelä et al). One function of HFE involves it binding to TfR (Figure 2) and thereby reducing the affinity of TfR for the Fe-Tf complex(Lebron et al, 1998). Since both HFE and TfR are membrane proteins, they are located in a very favorable position to bind.
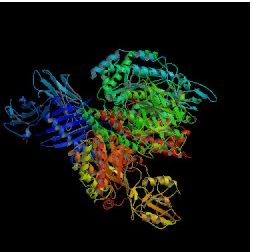
Image from Protein Data Bank.
Figure 2. HFE complexed with TfR. This binding reduces TfR's affinity for Tf by changing the configuration of TfR.
Experimental data has shown that TfR and Fe-Tf bind in a 2:2 ratio while TfR and HFE bind in a 2:1 ratio (Lebron et al., 1998). These data indicate that HFE and Fe-Tf have different binding sites on TfR. Thus, when HFE binds to TfR, it does not block Tf from binding but instead changes the conformation of the Fe-Tf binding site, which makes it more difficult for this group to bind. If Fe-Tf cannot bind to TfR, then Fe cannot enter the cell.
Furthermore, binding of HFE and TfR appears to be pH specific. That is, the molecules will bind at pH 7.5 but not pH 6.0 (Lebron et al., 1998). One possible explanation for this phenomenon is that the histidine cluster, which is neutral at pH 7.5, on the HFE protein (see Structure section) mediates the binding of these two molecules. When pH decreases to 6.0, the histidines become protonated, which causes HFE and TfR to dissociate. Since the pH of the cell membrane is 7.5, and the pH of the acidic vesicles is 6.0, the histidine cluster could act as a switch to regulate HFE binding to TfR (Lebron et al., 1998).
HFE decreases iron uptake without binding TfR
As discussed above, one definite function of HFE is inhibiting iron uptake into certain cells. However, data on how exactly HFE performs that function are conflicting. A more recent study indicates that HFE does not have to actually bind to TfR to regulate iron uptake. In this study, a mutated HFE (W81A HFE) that has a 5,000 fold decreased affinity for TfR was used. W81A HFE is not misfolded though and can still bind beta2m. Researchers found that cells with the mutated HFE and cells with the wild type HFE both accumulated 30% less iron that cells with a nonfunctional HFE (Figure 3) (Zhang et al., 2003). The same study also tested iron uptake when Tf was at a high enough level to block wild type HFE from binding to TfR. Again, cells with HFE had lower concentrations of Fe, even though their HFE was unable to bind TfR (Zhang et al., 2003). These results suggest that HFE can regulate iron concentrations in cells without necessarily inhibiting the interaction between Tf and TfR. Because a cascade of proteins are involved in iron uptake, processing, and efflux, HFE could interact with several different proteins throughout the process.
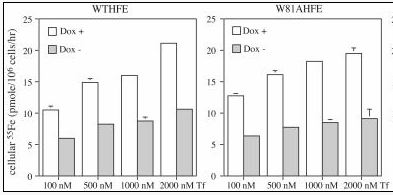
Image from Zhang et al., 2003. Permission pending.
Figure 3. Wild type and mutated W81AHFE both effectively regulate iron uptake. At increasing concentrations of Tf (nM), cells with either type of HFE in them had lower concentrations of Fe than cells without HFE. Approximately the same amount of Fe was present in cells with both wild type and mutated W81A HFE. Gray bars (Dox -) represent cells expressing HFE while white bars (Dox +) represent cells not expressing HFE.
HFE inhibits iron efflux
Although HFE inhibits iron uptake in some cell types, in others it inhibits iron exit (or efflux) from the cell. This function leads to an increased rather than decreased iron concentration in the cells. In a very recent study, researchers expressed HFE in a line of HT29 cells, a type of cell that behaves similarly to intestinal cells. A coprecipitation experiment found that HFE does interact with TfR in HT29 cells, even though iron levels were higher than normal in these cells (Davies and Enns, 2004). HT29 cells producing mutant W81A HFE (discussed above) also had higher than normal levels of iron, which implies that HFE need not interact strongly with TfR to fulfill its function. Although HT29 cells not expressing HFE absorbed iron at the same rate as wild type HFE and W81A HFE expressing cells, the cells not producing HFE exported iron 45% faster than either other type (Figure 4) (Davies and Enns, 2004). Thus, in at least HT29 type cells, HFE inhibits iron efflux. At least two proteins, FPN1 and ferroxidase HEPH, participate in iron export. When HFE is expressed, significantly less HEPH mRNA is produced (Davies and Enns, 2004) than in cells not producing HFE, which implies that HFE may inhibit iron efflux through decreasing levels of proteins necessary for export.
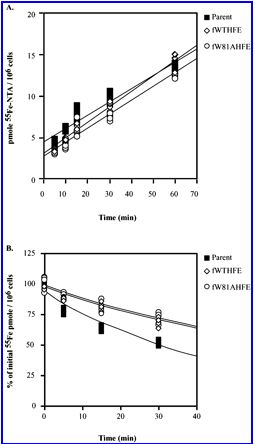
Image from Davies and Enns, 2004. Permission pending.
Figure 4. HT29 cells expressing HFE export Fe more slowly than cells not expressing HFE. The top graph shows that no difference exists between Fe uptake in the three cell types (no HFE, wild type HFE, W81A HFE). The bottom graph shows that that cells without HFE (black square) export iron 45% faster than cells expressing HFE.
Hereditary Hemochromatosis
Background
Type 1 hereditary hemochromatosis (HH) is the most common autosomal recessive disorder in people of Northern European descent (Lebron et al., 1998). In this disease, the body improperly stores and absorbs iron and ends up with too much iron in the liver, heart, and pancreas. This excess storage damages organs and can cause premature death if patients are not treated. The standard treatment is phlebotomy or bloodletting (Hemochromatosis, 2004). For more detailed information on the symptoms, diagnosis, and treatment of HH go to the National Digestive Diseases Information Clearinghouse.
Mutations
About 80% of people with HH have a single base pair mutation in the HFE DNA sequence that changes cysteine at the 260 position into tyrosine (C260Y) (Davies and Enns, 2004). This mutation prevents a disulfide bond from forming in the alpha3 region (discussed in Structure section) and also prevents HFE from binding to beta2m (Zhang et al., 2003). These structural disruptions result in the misfolding of the protein and prevent HFE from reaching the cell membrane. Since the disease is recessive, people must be homozygous for the mutation to have the disease phenotype.
A second, less common, mutation is one which changes the histidine at the 41 position to aspartic acid (H41A). While this mutation does not seem to affect the conformation of the entire protein, researchers believe that it does lead to a local rearrangement to avoid having two negative charges near each other (Lebron et al., 1998). This mutation alone does not lead to HH, but if a person is heterozygous for the C260Y mutation and also has the H41A mutation, they may develop the disease phenotype (Lebron et al., 1998).
References
Davies PS, Enns CA. 2004. Expression of the Hereditary Hemochromatosis Protein HFE Increases Ferritin Levels by Inhibiting Iron Export in HT29 Cells. Journal of Biological Chemistry 279 (24): 25085-25092.
Hemochromatosis. 2004 December. National Digestive Diseases Information Clearinghouse. http://digestive.niddk.nih.gov/ddiseases/pubs/hemochromatosis/index.htm. Accessed 14 February 2005.
Lebron JA, et al. 1998. Crystal Structure of the Hemochromatosis Protein HFE and Characterization of Its Interaction with Transferrin Receptor. Cell 93: 111-123.
Niemelä O, et al. HFE-Protein In Hereditary Hemochromatosis. http://cc.oulu.fi/~anatwww/hemochromatosis/info/. Accessed 14 February 2005.
Protein Data Bank. http://www.rcsb.org/pdb/cgi/explore.cgi?pid=133151108536448&page=0&pdbId=1A6Z. Accessed 10 February 2005.
The Hemochromatosis Gene. Gene Gateway - Exploring Genes and Genetic Disorders: A Web Companion to the Human Genome Landmarks Poster. http://www.ornl.gov/sci/techresources/Human_Genome/posters/chromosome/hfe.shtml. Accessed 14 February 2005.
Zhang A, et al. 2003. Mechanisms of HFE-induced regulation of iron homeostasis: Insights from the W81A HFE mutation. PNAS 100 (16): 9500-95005.
© Copyright 2005 Department of Biology, Davidson College, Davidson, NC 28036
Send comments, questions, and suggestions to: lumarcil@davidson.edu