This web page was produced as an assignment for an undergraduate course at Davidson College
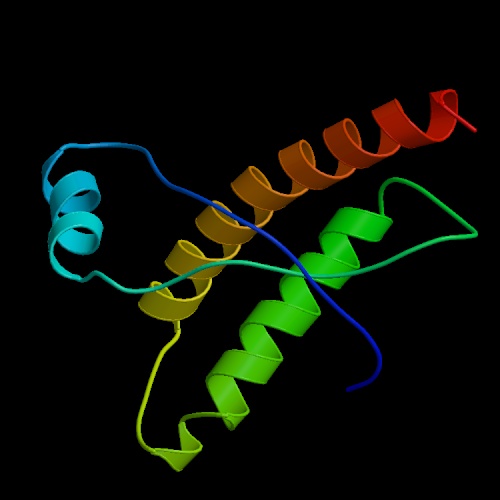
Prion Protein PrP
Image by Protein Data Bank
Prion Proteins: An Overview
Prion proteins, or PrPs, are thought to mediate copper ion (Cu 2+) recycling from synaptic clefts, thereby preventing oxidative damage to neurons due to excess Cu 2+. There is still some debate regarding their exact function, however it is known that the protein is expressed primarily in the central nervous system (CNS) (Roucou et al., 2005). An altered form of the normal prion protein, known as PrP Sc for PrP scrapie, has been found to cause fatal spongiform encephalopathies (SE) including Creutzfeldt-Jakob disease, mad cow disease and scrapie (DebBurman et al., 1997). These faulty PrP Sc prions convert normal PrP C prions into more PrP Sc , making them a class of unique infectious agents that multiply without a nucleic acid genome. Hence, these proteins were named prions meaning “protenaceous infectious particles” (DebBurman et al., 1997). An exploration of the function of normal prion proteins and the conformational changes that convert them to infectious agents will give scientists a greater understanding of SEs and perhaps an opportunity to discover a cure.
Function of PrP C
Cellular prion protein, PrP, provides neuroprotection through copper homeostasis, antioxidant activities, and the inhibition of cell apoptosis (Roucou et al., 2005). PrP is a glycoprotein tethered to the cell surface via a glycosyl phosphatidyl inositol (GPI) anchor at the protein’s C terminus (Jones et al., 2004). The presence of free floating PrP in the cytosol indicates that it may also have intracellular functions (Roucou et al., 2005).
Studies indicate that PrP C plays a role in copper homeostasis within the cell as elevated synaptic Cu 2+ levels are shown to promote PrP C mediated Cu 2+ endocytosis (Jones et al., 2004). PrPs control Cu 2+ concentrations within lymphoid or nerve cells by binding extracellular and intracellular Cu 2+ ions in their octarepeat regions (Jones et al., 2004). PrP C expression in cells was also found to increase antioxidant enzyme activities (Roucou et al., 2005). PrP is also thought to prevent apoptosis through inhibition of the mitochondrial apoptotic pathway.
The role of PrP in cell survival was examined by Roucou et al. (2005) who found a conserved sequence homology between the BH2 domain of the Bcl-2 protein and the octarepeat region of PrP. Bcl-2 protein suppresses cell death by homodimerizing with Bax protein, thereby inhibiting Bax function. Bax protein induces cell death through the apoptotic pathway by permeating, and thus destroying, the mitochondrial membrane (Roucou et al., 2005). Roucou et al. found that coexpression of both cystolic and membrane-bound PrP with Bax abolished Bax-mediated cell death (Figure 1). They were led to conclude that PrP, like Bcl-2, was involved in antiapoptotic activity.
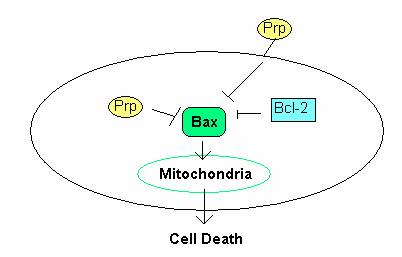
Figure 1: The role of PrP in the prevention of cell death. PrP C prevents apoptosis through inhibition Bax in the mitochondrial apoptotic pathway. Figure adopted from Fig. 2 in Roucou et al., 2005.
Prion Proteins: Structure and Location of PrP
The gene that encodes PrP is located on human chromosome 20. The transcription length of the gene is 855 bp long and includes 2 exons. Translation of the gene encoding PrP yields a protein with 217 residues, or amino acids, and an overall charge of 11 (Sanger Institute- Ensembl Protein Report, 2005). Once transcribed, the PrP protein can adopt one of 2 conformations. Both conformations, PrP C and PrP Sc , have the same primary structure, or amino acid sequence, but differ in their secondary and tertiary structures. The amino acid sequence of the PrP protein is shown in figure 2 (Sanger Institute- Ensembl Protein Report, 2005).

Figure 2: Amino Acid Sequence of the gene encoding PrP . Red highlights represent non-synonymous single nucleotide polymorphisms (SNPs). Green highlights represent synonymous SNPs. Blue highlights represent insertions and deletions. Orange letters represent the octarepeat region. Image by Sanger Institute- Ensembl Protein Report, 2005.
Primary Structure
An analysis of the primary structure of PrP gives insight into its function. As aforementioned, PrP binds Cu 2+ in order to mediate copper ion recycling within the cell. A recent study by Jones et al. (2004) shed light on the mechanisms by which Cu 2+ binds to PrP through analysis of the N and C terminal domains of the PrP protein (Figure 3).
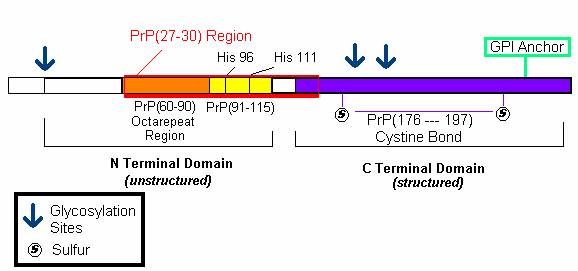
Figure 3: The amino acid sequence of Mature PrP. The N terminal domain consists of an octarepeat region (PHGGGWGQ) and encodes His 96 and 111 copper binding sites. The C terminal domain contains the sequence for the GPI anchor and the regions connected by cystine bonds. Both regions contain post-translational glycosylation sites.
It was previously thought that PrP protein bound 4 copper molecules with increasing affinities to its octarepeat region ( PHGGGWGQ) (Jones et al., 2004) . A study by J ones et al. (2004) revealed that copper in fact binds more tightly to the PrP(91-115) domain than the octarepeat region. More specifically, they found that Cu 2+ ions bound to HIS 96 and HIS 111 regions within the PrP(91-115) domain. They speculate that this domain may act as a copper dependent hinge whereby copper first binds to HIS regions which then activate the octarepeat regions to bind and transport copper as needed (Jones et al., 2004). The PrP(91-115) domain, with HIS 96 and 111 regions, is thought to be the domain that misfolds to yield the deadly PrP Sc conformation of the prion protein (Jones et al., 2004).
Secondary Structure
PrP C, the conformation which occurs in healthy, non-infected cells, is composed of 3 alpha helices and 2 beta-pleated sheets (DebBurman et al., 1997). Although the structure of PrP Sc has not yet been ascertained, it is predicted that it is composed of primarily beta pleated sheets as opposed to alpha helices (Figure 4) (DebBurman et al., 1997). A study by Horwich et al. (1997) determined that the PrP C conformation is composed of approximately 40% ![]() -helical content with little or no
-helical content with little or no ![]() sheet content, while the PrP 27–30 form contains approximately 50%
sheet content, while the PrP 27–30 form contains approximately 50% ![]() sheet and only 20%
sheet and only 20% ![]() -helical (Figure 4). It is thought that one of the alpha helices in PrP C is misfolded and converted to a beta sheet in PrP Sc (Horwich et al. 1997).
-helical (Figure 4). It is thought that one of the alpha helices in PrP C is misfolded and converted to a beta sheet in PrP Sc (Horwich et al. 1997).
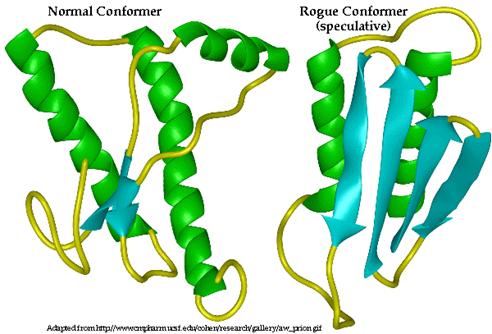
Figure 4: Three-dimensional structure of PrP C (left) and proposed 3D structure of PrP Sc (right). Alpha helices are indicated in green while beta sheets are indicated in blue. PrP C is composed primarily of alpha helices while PrP Sc is composed primarily of Beta pleated sheets. Permission Pending from BSE Information at UIUC at http://w3.aces.uiuc.edu/AnSci/BSE/.
Of the three alpha helices of PrP C, two of them are longer than the third, indicating that this third is intrinsically less stable due to the decreased number of hydrogen bonds in the coil. Consequently, it is thought that this more unstable, shorter alpha helix, may be that which is converted to the beta sheet (DebBurman et al., 1997). Between two of the alpha helices of PrP C, there is evidence of a single cystine bond (cys- S-S- cys) (McCusker et al., 2005). These bonds are typical of secreted proteins and those located near the plasma membrane, as is PrP. This cystine bond is thought to stabilize the two alpha helices that it spans in PrP C (McCusker et al., 2005). However, in the PrP Sc conformation, this cystine bonds is though to be broken, destabilizing the protein and exposing the unbound sulfurs on the cystine complex towards the outside of the protein (McCusker et al., 2005). This loss of the cystine bond is thought to be a consequence of 1) a mutation in the gene encoding normal PrP C, 2) random misfolding of a PrP C protein or 3) a result of the interaction of PrP C with PrP Sc , among others.
Prion Proteins: Agents of Infectious Disease
Summary: PrP C vs. PrP Sc
PrP C |
PrP Sc |
primarily beta sheet structure |
|
protease susceptible |
resistant to protease |
monomeric species |
forms multimeric aggregates |
stable monomer |
less stable monomer, self propagates for stability |
normal resistance |
extremely resistant to heat, chemicals, radiation and strong solvents
|
detergent soluble |
detergent insoluble |
Prion Disease: An Overview
The process by which PrP Sc causes spongiform encephalopathies can be summarized in four steps (Figure 5):
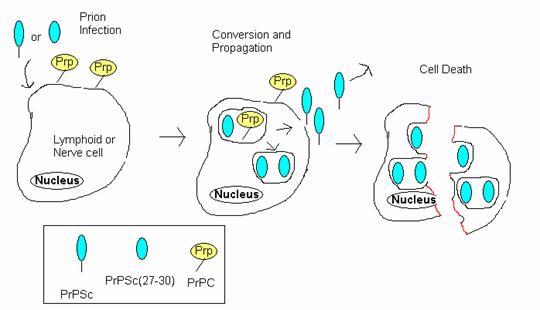
Figure 5: Prion Infection Resulting in cell death. This diagrams the process by which prions infect cells and ultimately cause fatal spongiform encephalopathies. PrP Sc first enters the cell and converts normal PrPs into more PrP Sc . These newly created PrP Sc molecules either exit the cell to infect other cells or accumulate in the cell, ultimately causing the cell to lyse. Adopted from BSE Information at UIUC at <http://w3.aces.uiuc.edu/AnSci/BSE/Index_BSE_Info_at_UIUC_Science.htm> under 'Cell conversion'.
Prion Infection
Faulty PrP Sc conformations may enter the body through consumption of scrapie-infected cells or are produced as a consequence of mutations in the gene encoding normal PrP C. In humans, mutations within the Human PrP gene cause Creutzfeldt-Jakob disease (CJD), Gerstmann-Straussler-Scheinker disease, and Fatal Fetal Insomnia (DebBurman et al., 1997). PrP Sc can also enter the body through the consumption of meat of cows infected with mad cow disease or sheep infected with scrapie (DebBurman et al., 1997).
Conversion of PrP C to PrP Sc
The conversion of PrP C to PrP Sc is thought to be mediated by direct interactions between PrP C and PrP Sc . This is evidenced by mice PrP-/- knockout studies yielding mice resistant to scrapie infection (Roucou et al., 2005). Scientists speculate that PrP-/- knockout mice were viable because the role of PrP is so important that its function in neuroprotection is redundant (Roucou et al., 2005). In support of these findings, studies of chaperone mediated PrP to PrP Sc conversion demonstrate that while some chaperones could induce transformation of PrP C, none could promote conversion in the complete absence of PrP Sc (DebBurman et al., 1997). Two chaperones thought to mediate this conversion are GroEL and Hsp 104 (heat shock protein 104) (DebBurman et al., 1997).
Attack by Proteinases
PrP Sc (27-30) is created when proteinases within the cell, which are made to destroy foreign proteins, attempt to eradicate PrP Sc through digestion but instead only truncate it (Zanusso et al., 2004). Indeed protease resistance is one of the primary characteristics of PrP Sc protein and is used as a diagnostic tool for cells that are thought to be scrapie-infected. Whereas normal PrP C is sensitive to proteinase K digestion, PrP Sc loses only 50-80 amino acids in its N terminus and another 70-80 amino acids in its C terminus (Zanusso et al., 2004).
Formation of amyloid plaques
Once created, this protease resistant core, with its compact size and instability, aggregates with other PrP Sc (27-30) fragments to form aggregations called amyloid plaques. Recall that in PrP Sc , a normally intact cystine bond is disrupted, yielding exposed and unstable sulfur atoms that orient themselves outward from the protein (McCusker et al., 2005). Exposed sulfurs on PrP Sc (27-30) fragments bind to one another creating growing disulfide polymers within the cell (Figure 6). Ultimately, the cells lyse, releasing more PrP Sc into extracellular spaces much like viruses in their lytic cycles.
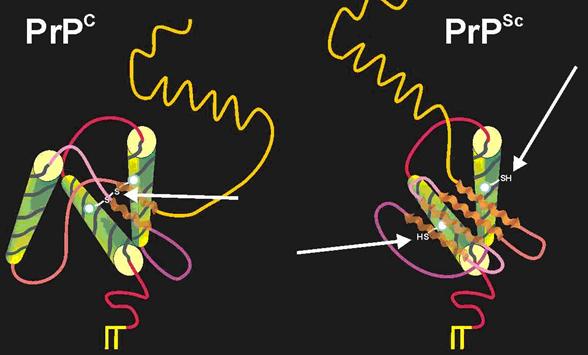
Figure 6: Disruption of Cystine bond in Conversion from PrP C to PrP Sc . Between two of the alpha helices of PrP C, there is evidence of a single cystine bond (cys- S-S- cys). However, in the PrP Sc conformation, this cystine bonds is though to be broken, destabilizing the protein and exposing the unbound sulfurs on the cystine complex towards the outside of the protein. Permission Pending from BSE Information at UIUC at http://w3.aces.uiuc.edu/AnSci/BSE/Index_BSE_Info_at_UIUC_Science.htm under '3D structure'.
Links
http://www.receptors.org/Prion/htmls/entries.html A list of all prion proteins available in the Protein Data Bank.
http://www.mad-cow.org/ The Official Mad Cow Disease Homepage
Works Cited
DebBurman, S., Raymond, G., Caughey, B., and S. Lindquist. 1997. “Chaperone-supervised conversion of prion protein to its protease-resistant form.” Proceedings of the National Academy of Science USA. 94: 13938-13943.
Horwich, A. and J. Weissman. 1997. “Deadly Conformation- Protein Misfolding in Prion Disease.” The Cell 89(4): 499-510.
Jones, C. E., Abdelraheim, S., Brown, D., and J.H. Viles. 2004. “Preferential Cu 2+ Coordination by His 96 and His 111 Induces B-Sheet Formation in the Unstructured Amyloidogenic Region of the Prion Protein.” Journal of Biological Chemistry. 279(31): 32018-32027.
McCusker, Robert, et al. 2005. Bovine Spongiform Encephalitis Information Online. Available <http://w3.aces.uiuc.edu/AnSci/BSE/> Accessed February 11, 2005.
Roucou, X. and LeBlanc, A. 2005. “Cellular Prion protein neuroprotective function: implication in prion diseases.” Journal of Molecular Medicine. 83: 3-11.
Sanger Institute, 2005. Ensembl Human Genome Server: PNRP. Available <http://www.ensembl.org/Homo_sapiens/geneview?gene=OTTHUMG00000031786&db=vega> Accessed February 10, 2005.
Zanusso, G., Farinazzo, A., Prelli, F., Fiorini, M., Gelati, M., Ferrari, S., Righetti, P., Rizzuto, N., Frangione, B., and S. Monaco. 2004. “Identification of Distinct N-Terminal Truncated Forms of Prion Protein in Different Creutzfeldt-Jakob disease Subtypes.” Journal of Biological Chemistry. 279(37): 38936-38942.
Questions or Comments? Please e-mail Katie Winter