|
C O N P S |
These Chime images provided by the Protein Data Bank depict the 3D crystal structures of the globular domains of sheep (residues 121-231 ) and human (residues 119-226) prion proteins, in normal and dimer forms, respectively. The proteins are non-glycosylated and lack portions of the N-terminus, including the hydrophobic leader sequence and octarepeat regions, and the GPI anchor of the C-terminus. Because prion is a membrane bound protein, a fraction of the sequence was likely removed in order to extract the protein for crystallography.
Click the buttons to enjoy this interactive Chime tutorial:
To reset Sheep Prp. Click
here.
To reset Human PrP. Click
here.
Structure:
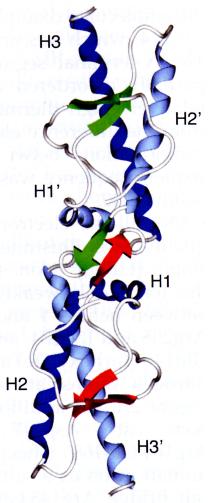
>Let's look first at sheep Prp in its normal form. As aforementioned, this molecule consists of 3 alpha helices (type H) and 2 anti-parallel beta-pleated sheets. Observe alpha helix 1 (H1) , alpha helix 2 (H2) , and alpha helix 3 (H3) of human prion protein. Observe G1 . There are also two shorter type-G helices- G1 and G2 - whose loop diameters are significantly smaller than type H helices. Here are the anti parallel beta sheets B1 & B2. .
Notice that these helices together with the beta sheets constitute a single compact and highly stable core. As such, prion carries out its normal duties mediating copper ion concentration within the cell & synaptic clefts. When cells become infected with PrP scrapie (PrP Sc), however, normal prion undergoes conformational changes which destabilize the molecule and allow for the aggregation of amyloid plaques. Let's explore these conformational changes by examining human prion protein in dimer form.
> Human and sheep prion proteins have the same secondary structures (recall also that sequence homology between prion orthologs is highly conserved). Again, here are alpha helices H1 , H2 , and H3 of human prion protein. Observe G1 , G2 , and the anti parallel beta sheets B1 & B2. .
Notice that prion in its dimer form is highly unstable. The most apparent reason for this change is the loss of the cysteine bond, which stabilizes H2 and H3 of normal prion , but is lost during the conversion of PrP C to PrP Sc . Notice that the exposed and unbound sulfurs face outward. Soon we'll see how the loss of this stabilizing cysteine bond leads to prion oligomerization.
Normal prion protein is also more inherently stable due to the many water molecules (in blue), some partially buried, that help stabilize the conformation . Notice that in the dimer form of human prion the water molecules are unevenly distributed towards one area of the protein, leaving H3- which lacks the stabilizing water molecules-highly unstable .
For both molecules, hydrogen bonds increase protein stability, however slightly more H bonds are present- and of shorter length- in sheep PrP than in human dimer PrP (especially in areas that lack alpha helices or beta sheets).
Oligomerization:
Haire et al. (2004) proposed a mechanism for the oligomerization of normal prion protein. The primary conformational change is the destruction of the cysteine bond that bridges H2 and H3. Haire et al. hypothesized that after disruption of this disulphide bond, H3 is exchanged between two PrP molecules, followed by the annealing of the disulphide bonds between the cysteines of the newly aligned PrPs (Figure 1).
Figure1. From Figure 2C of Haire et al. (2004) Permission Pending.
As a result of this oligomerization, the structures of the helices and the local interactions between each of the monomers is preserved. In other words, PrP Sc monomers are 'encouraged' form dimers because they can regain the stability that was lost after the disruption of their cysteine bonds (Haire et al., 2004).
Notice in Figure 2C the addition of an anti-parallel pair of beta-sheets (with each PrP monomer providing 1 beta-sheet). These residues (188-200) normally compose the C terminal of H2. Interestingly enough, these residues consist primarily of threonines, whose properties and placement lower the stability of H2 at the C terminal . As such, these threonines have the propensity to undergo an alpha to beta transition (recall that PrP Sc is thought to be composed of more beta character than PrP C). Note also that these 4 consecutive threonines are highly conserved among prion orthologs (Figure 2).

Figure 2. Residues 188-201 of prion orthologs. Notice the high degree of threonine composition in this segment.
Notice that in the dimer conformation that the alpha helix is lost and stretched almost flat, these are the beginnings of beta sheet formation . Notice also the loss of hydrogen bonds corresponding to this transformation- in sheep PrP & in human PrP in dimer form . Haire et al. (2004) proposed that that the partial unwinding of H2 at this end triggers the alpha to beta sheet transition.
So far, we've discussed the ways in which two prion monomers can combine to form a dimer. This is just the beginning of oligomerization. Once a dimer is formed, two dimers can aggregate to form a tetramer, and so on. The dimers combine by 'stacking' themselves upon each other. During this process, the anti-parallel beta sheet pairs are transformed into 4 stranded intermolecular beta-sheets (Figure 3).
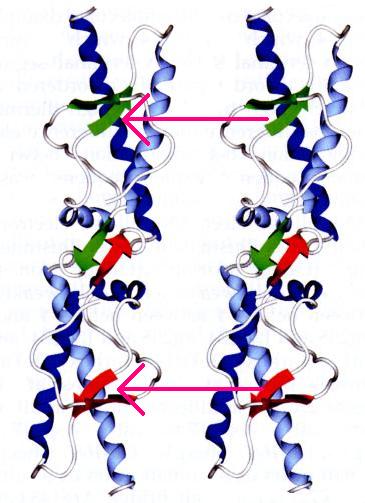
Figure3. Adopted from Figure 2C of Haire et al. (2004) Permission Pending.
According to Haire et al. (2004), the most significant contact between the beta sheet pairs occures at Met129 residues. Let's examine methionine in normal and dimerized PrP. Here is where Met129 is normally positioned in sheep PrP . In dimerized PrP, however, Met 129 is inverted and faces outward- leaving it in prime position to make contact with other Met 129 residues on adjacent dimers .
Recall that in our study of orthologs, this region is highly conserved across species. The importance of Me129 in oligomerization explains why mutations at this residue may result in barriers to PrP Sc transmission. Recall, for example, that individuals heterozygous (V/M) at residue 129, had decreased risk of Alzheimer's and other amyloid plaque-related diseases as compared with homozygous individuals (M/M). (For more information see the previous section on 'Prion orthologs').
Function:
Let's now take a minute to explore how conformational changes that convert normal PrP to dimer form also adversely affect a prion protein's normal function. As you recall PrPs control Cu 2+ concentrations within lymphoid and nerve cells by binding extracellular and intracellular Cu 2+ ions in their octarepeat regions (Jones et al., 2004). It thought that these octarepeat regions are first activated by the binding of copper to HIS 96 and HIS 111 regions. Although these pdb files lack residues comprising the octarepeat regions, HIS 96 and HIS 111 regions are visible.
In normal PrP HIS 96 and HIS 111 regions span H1 and face in roughly the same direction . Notice however, in dimer form, H1 flips and twists upward resulting in the disorientation of HIS 96 and HIS 111 copper dependent hinges . This suggests that the normal function of copper recycling is likely impaired during the conformational change from PrP C to PrP Sc.
Epitopes (Protein X):
The conversion of PrP to PrP Sc is not thought to occur spontaneously. Kaneko et al. (1997) demonstrated that an unknown chaperone protein, named protein X, participates in the conversion of PrP C to PrP Sc. Kaneko et al. (1997) proposed that protein X binds to epitopes (shown here ) on PrP C forming a heterologous complex that then interacts with other PrP Sc proteins. Two chaperone proteins thought to mediate this conversion are GroEL and Hsp104 (DebBurman et al., 1997). After PrP C to PrP Sc conversion, protein X dissociates due to its lack of affinity for PrP Sc. This lack of affinity is likely due to the separation and disorientation of the epitopes are oriented prion in dimer form .
Kaneko et al. (1997) also found that PrP C proteins with mutated epitopes could act as 'dominant negatives' in the inhibition of PrP Sc amyloid plaque formation. The two proposed mechanisms for this inhibition are as follows: 1) epitope mutations prevent the initial binding of PrP C or 2) protein X binds to PrP C, but cannot dissociate from it after interaction with other PrP Scs.
References:
DebBurman, S., Raymond, G., Caughey, B., and S. Lindquist. 1997. “Chaperone-supervised conversion of prion protein to its protease-resistant form.” Proceedings of the National Academy of Science USA. 94: 13938-13943.
Haire, L.F., Whyte, S., Vasisht, N., Gill, A., Verma, C., Dodson, E., Dodson, G., and P. Bayley. 2004. Journal of Molecular Biology. 336: 1175-1183.
Jones, C. E., Abdelraheim, S., Brown, D., and J.H. Viles. 2004. “Preferential Cu 2+ Coordination by His 96 and His 111 Induces B-Sheet Formation in the Unstructured Amyloidogenic Region of the Prion Protein.” Journal of Biological Chemistry. 279(31): 32018-32027.
Kaneko, K., Zulianello, L., Scott, M., Cooper, C., Wallace, A., James, T., Cohen, F., and S. Prusiner. 1997. "Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation." Proceedings of the National Academy of Science. 94: 10069-10074.
Knaus, K.J., Morilla, M., Swietnicki, W., Malone, M., Surewicz, W., and V. Yee. 2001. "Crystal Structure of the human prion protein reveals a mechanism for oligomerization." Nature Structural Biology. 8: 770-774.