Review Paper
Creating Bacterial Strains from Genomes That Have Been Cloned and Engineered in Yeast
Lartigue, C, S Vashee, MA Algire, R Chuang, GA Benders, L Ma, VN Noskov, EA Denisova, DG Gibson, N Assad-Garcia, N Alperovich, DW Thomas, C Merryman, CA Hutchison III, HO Smith, JC Venter, and JI Glass
Science 2009, 325: 1693-1696
Summary of data presented
The laboratory that produced this paper is in the process of constructing a living microbe from synthetic building blocks. Already, Lartigue et al. have transplanted the Mycoplasma mycoides genome into the cellular environment of Mycoplasma capricolum. Additionally, the genome of Mycoplasma genitalium has been chemically synthesized using several techniques and species. First, in vitro assembly reactions were used to piece small stretches of the genome together. Next, larger pieces of the genome, up to a quarter of the entire genome in length, were cloned in E. coli. Finally, because E. coli is not large enough to handle larger segments of DNA, homologous recombination in the yeast, Saccharomyces cerevisiae, was used to complete the synthesis of the M. genitalium genome. The next step in generating a synthetic living organism is to isolate this synthetic genome and transplant it into a cellular environment capable of sustaining life given this species’ genome.
In this study, both a natural and yeast synthesized version of the M. mycoides genome is transplanted into the cellular environment of M. capricolum. These two species have a faster growth rate than M. genitalium making them more convenient for this study. The first step taken to alter the M. mycoides genome was to transform the bacteria with a vector containing a selectable marker for antibiotic resistance and the β–galactosidase gene. These selectable markers will allow the scientists to determine which bacteria have successfully obtained transplanted M. mycoides genomes after transplantation. In addition to selectable markers, the vector contained yeast sequences that made reproduction in yeast possible for the M. mycoides genome. Lartigue et al. sequenced the genome after transformation to ensure that the entire vector was incorporated into the genome although data was not shown for these findings. After transformation, the M. mycoides genome is referred to as YCpMmyc1.1 for the remainder of the paper. After altering the M. mycoides genome, YCpMmyc1.1 was isolated from bacterial cells and transformed into yeast spheroplasts from two different strains of yeast: VL6-48N and W303a. A spheroplast is a cell, in this instance a yeast cell, that has been altered so that there is a partial loss of the cell wall and increased osmotic sensitivity. To check that the entire genome was effectively transformed, two methods were used. First, multiplex PCR was performed. Multiplex PCR uses multiple primers to amplify multiple pieces of DNA. These PCR products were distinguished using clamped homogenous electric fields (CHEF) gel electrophoresis. In this form of electrophoresis, very large (up to 5Kb) pieces of DNA can be effectively separated from one another. The data from these experiments is shown in supplemental figures only, but according to the authors, all colonies screened seemed to contain complete YCpMmyc1.1 genomes. Based on this finding, the authors concluded that the M. mycoides genome is stable in yeast.
Once the YCpMmyc1.1 genome was transformed into the two yeast strains, Lartigue et al. engineered a change to the genome. First, the Type III restriction endonuclease (typeIIIres) gene open reading frame was deleted from the genome by introducing a knockout cassette. Basically, a piece of DNA with homologous regions specific to either end of the typeIIIres gene was introduced in the yeast. Contained within this region of DNA were a URA3 marker and the SCEI endonuclease gene, which was under the control of the GAL1 promoter. The URA3 marker was used as a selectable marker. The introduction of the SCEI endonuclease gene allowed the scientists to turn transcription of this gene on and off using the GAL1 promoter. When the SCEI endonuclease was activated, it caused this restriction enzyme to cut the DNA at a site near the beginning of the GAL1 promoter. After the DNA is cleaved, there are two regions of tandem repeat sequences in this area of the genome that may reanneal after the cut occurs effectively deleting the knockout cassette from the genome.
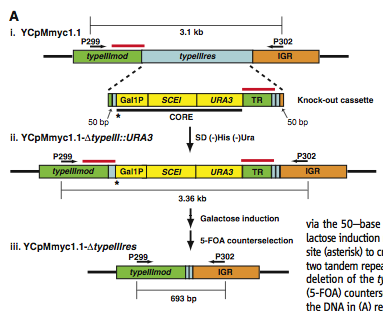
Figure 1A
Figure 1A shows schematics of this process. In Figure 1 i., a 3.1kb portion of the YCpMmyc1.1 genome is displayed. Within this portion of the genome are the typeIIImod region, the typeIIIres gene, and an intergenic region or (IGR). Directly beneath this schematic is a model of the knock-out cassette. The two homologous regions on either end of the cassette have dotted lines drawn connecting them to the regions of homology in the YCpMmyc1.1 genome. The GAL1 promoter is shown with the SCEI endonuclease gene directly to the right of it. Next, the URA3 selectable marker is shown. Finally, an area of tandem repeats (TR) is shown. After selection for yeast that can grow in media lacking His and Ura, Figure A ii. displays what the first mutant genome looks like. Homologous recombination causes the typeIIIres open reading frame to be replaced with the knockout cassette. This version of the genome with the knock-out cassette is referred to as YCpMmyc1.1-ΔtypeIII::URA3. Areas of tandem repeats are highlighted with a red line above the DNA. The star shows the cut site from the SCEI endonuclease. Finally, after galactose induction, the cassette will be deleted completely resulting in YCpMmyc1.1-ΔtypeIIIres (Figure 1Aiii). These mutants are selected using 5-flouroorotic acid (5-FOA) counterselection. Growing yeast in medium containing 5-FOA will kill any yeast containing the URA3 gene. The typeIIIres open reading frame and knockout cassette have been removed completely resulting in a seamless deletion of typeIIIres open reading frame.
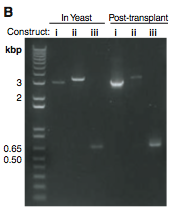
Figure 1B
In Figure 1B, the changes to the genome are confirmed using PCR. The PCR primers are shown in Figure 1A as arrows labeled P299 and P302. From this schematic, you can see that construct YCpMmyc1.1-ΔtypeIII:URA3 should be the largest followed by normal YCpMmyc1.1, and the smallest construct should be YCpMmyc1.1-ΔtypeIIIres. PCR confirms that these PCR products are the expected size both in yeast as well as post-transplant cells.
Next, Lartigue et al. attempted to transform YCpMmyc1.1 from yeast to M. capricolum cells. No colonies were recovered from this transplantation, however. The scientists hypothesized that this was due to native endonucleases from the M. capricolum cells cleaving the introduced genome. To overcome this obstacle, two methods were utilized. First, the only restriction enzyme found in M. capricolum was inactivated by inserting a selectable marker directly into the coding region of the gene. This cell line was referred to as M. capricolum RE(-). Secondly, the scientists used three different methods to methylate the YCpMmyc1.1 genome. Methylation protects DNA from degradation caused by restriction enzymes. The methods used to methylate the DNA were in vitro methylation using M. capricolum or M. mycoides extracts or by in vitro methylation using methylases purified from M. mycoides.
Table 1
Table 1 summarizes the results of several transformation studies. Bacterial colonies were selected based on their resistance to the antibiotic tetracycline. Additionally, since the transplanted colonies should contain β–galactosidase, only blue colonies represented transformed colonies. The chart is organized based on yeast strain from which the YCpMmyc1.1 genome was isolated. For the VL6-48N strain, the YCpMmyc1.1 genome was given several different methylation treatments. The genome was either not methylated, methylated by one of three different methods, or mock-methylated. Secondly, the methylated or unmethylated genome was transplanted into either wildtype M. capricolum cells or M. capricolum cells with a malfunctional restriction enzyme (M. capricolum RE(-)). Note that when the genome was not methylated or mock-methylated and transplanted into wildtype cells, no transplantation occurred. However, in every other instance, colonies were recovered. For genomes isolated from W303a strains, none of the genomes were methylated but all were transplanted into M. capricolum RE(-) cells. In this experiment, different forms of the genome were transplanted including YCpMmyc1.1, YCpMmyc1.1-ΔtypeIIIres::URA3, and YCpMmyc1.1-ΔtypeIIIres. Additionally, one of the clones from the knockout cassette transformation had a 500kb deletion when screened. This mutant genome was used as a negative control since the deletion interrupted genes essential to life. When YCpMmyc1.1-Δ500kb was transplanted, no transplants were recovered. These different genomes that have been transplanted will be used later in the paper to prove that the M. mycoides genome synthesized in yeast was specifically transplanted into the M. capricolum cells and to show that genetic engineering can be performed to bacterial genomes while contained within yeast cells. These results demonstrate that avoiding interference with host cell restriction enzymes is essential to a successful transplant. Additionally, the scientists argue that it did not matter whether the YCpMmyc1.1 genome was purified away from yeast genomic DNA prior to transplantation because colonies were recovered regardless of digestion of yeast genome prior to transplantation.
Figure 2A
Next, Lartigue et al. verified that the colonies recovered were from M. mycoides using Southern blots. The probe used for this study was an element of the M. mycoides genome specific to this species. DNA from various cell types with either transplanted genomes or native genomes were digested with Hind III restriction enzyme and separated by gel electrophoresis prior to being hybridized with the probe. Notice in Figure 2A that the wildtype M. capricolum cells do not hybridize with the probe while all transplanted cells (YCpMmyc1.1, ΔtypeIIIres::URA3, and ΔtypeIIIres) do. There is also a positive control for native M. mycoides cells in lane 2 of this blot.
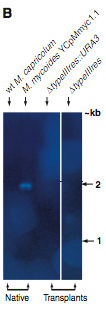
Figure 2B
In this figure, genomic DNA from either native M. capricolum or M. mycoides or transplanted ΔtypeIIIres::URA and ΔtypeIIIres was digested with Eco RV and separated by gel electrophoresis. A Southern blot was performed using a probe specific to the typeIIIres gene sequence. Notice that this gene is only present in native M. mycoides, but not native M. capricolum or either transplanted genome shown.
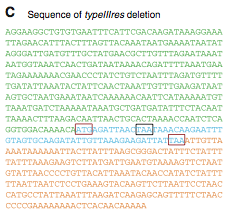
Figure 2C
Lartigue et al. sequenced the YCpMmyc1.1-ΔtypeIIIres M. mycoides genome transplanted into M. capricolum cells. This sequence analysis shows the deletion of the typeIIIres gene. The start and stop codons of the typeIIIres gene are boxed in red. The stop codon of the typeIIImod gene is boxed in black.
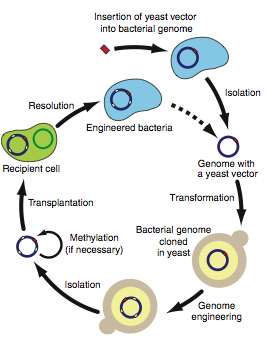
Figure 3
In the final figure, Lartigue et al. present a schematic of the new method presented in this paper. The data presented in this paper have shown that it is possible to engineer a bacterial cell by altering its genome after transformation of the entire genome into yeast. Figure 3 shows the steps necessary for this process. First, a yeast vector must be inserted into a bacterial genome. Next, isolate the bacterial genome and transform it into yeast. Once within yeast, any yeast genetic manipulation methods available may be utilized to alter the bacterial genome. After genome engineering has taken place, you can again isolate the bacterial genome and methylate to protect it if necessary prior to transplanting it into a recipient cell. Resolution removes the native genome from the transplant host cell. This cycle can be repeated multiple times to induce new traits into a species. Therefore, this method could be used to create new strains of bacteria that could not have been engineered using only bacterial genetic manipulation.
Critique
When testing the transformed YCpMmyc1.1 genome, the authors used a method that did not check the entire genome for sequence variation. The use of multiplex PCR and CHEF gel electrophoresis could check for major deletions but not changes between amplified sequences of DNA nor point mutations. I think it was pre-emptive to assume at this point in the paper that the M. mycoides genome is stable in yeast. It is possible that small mutations were introduced during the transformation process that could have large effects on the genome. For instance, if point mutations were introduced in essential genes, then attempts to transplant the genome into a new bacteria species could completely fail due to these alterations to the genome. Clearly, because the scientists were able to transform and further sequence the resulting genome without major changes to YCpMmyc1.1, it later becomes clear that these potential smaller mutations to the genome were not a problem.
I think the design of the knockout cassette is quite creative and useful. The cassette contains a selectable marker as well as a gene for a restriction enzyme under the control of the GAL1 promoter. The selectable marker ensures that you can easily isolate cells that have been transformed. Once you activate the GAL1 promoter, the restriction enzyme, SCEI, is transcribed. This protein will cleave the cassette near the beginning of the GAL1 promoter. This cleavage promotes the interaction of two tandem repeat sequences. One of these repeat sequences is present in the native genome prior to the typeIIIres open reading frame while the other is at the end of the knockout cassette. The interaction of these tandem repeats can cause homologous recombination which will completely remove the knockout cassette. Next, by selecting for cells without the URA3 protein, you can isolate only cells that have deleted the cassette. This is a very intricate method used to create seamless deletion of typeIIIres.
For the claim that yeast genomic DNA does not affect transformation, I have a problem with the data presented. First, they only test the effect of yeast genomic DNA for one type of methylated sample in wild type cells. Second, there seem to be significantly less colonies when genomic DNA is removed. Since no p value is reported, I cannot draw any conclusions. Also, I think they should have tried to remove yeast genomic DNA under multiple conditions. At the very least, testing the effect of yeast genomic DNA when transplanting untreated YCpMmyc1.1 into M. capricolum RE (-) should have been done since this is the method of transplantation used most frequently in this paper.
For Figure 2A, I am concerned that lane 1 shows nothing. Perhaps the scientists could have also included a probe specific to some regulatory protein found in both versions as a loading control to ensure that DNA was in fact loaded in lane 1. Also, for Figure 2B, only one band is present in one lane. A loading control could have helped us compare lanes to ensure that DNA was present. Also, the transplanted YCpMmyc1.1 genome could have been included to show that the typeIIIres gene was present. It is another good positive control to prove that the yeast genetic engineering employed in this study was effective.
Figure 3 contains the only reference to the host cell native genome in the entire paper. When you transplant the M. mycoides genome, the M. capricolum genome is still present. Since no attempt was made to remove the M. capricolum genome, it is quite possible that the cell would not survive if the native genome was replaced by the M. mycoides genome. I think it is odd that this paper shows the ‘reconstitution’ step where the native genome is removed but offers no method by which it could be degraded in the text.
Although I have a few concerns about this paper, I think that, overall, well controlled, well designed experiments were utilized that prove it is possible to transplant bacterial genomes altered in yeast into a different species of bacteria. Furthermore, with further refining to these methods, this process will probably become a very useful tool to the study of bacterial pathogenesis.
Future Studies
My first concern that ought to be addressed in future studies is the presence of native bacterial genome in host cells used for transplantation. It is necessary to remove the native genome either before or after transplantation in order to ensure that the transplanted genome can sustain life on its own. Additionally, in order to use this method to produce new bacterial strains, the removal of this native genome is essential. It would probably be difficult to remove the native genome after the transplanted genome is introduced since the two genomes are so similar. The only thing that would concretely set the two apart would be the remaining yeast components in the engineered genome used to ensure this genome can replicate in the yeast host. If there was a way to only protect the chromosome containing these yeast components while degrading the other DNA, this tool could be used to get rid of the native genome. Otherwise, it may be more efficient to remove the native genome prior to transplantation. However, I do not know how long a cell could survive or if it could survive after its genome is removed. A method that could be used to remove the genome would be to add nucleases to degrade the DNA present in the cell. Of course these nucleases would have to be neutralized prior to the addition of the transplanted genome.
In order to test that yeast synthesized genomes transplanted into bacterial cells lacking native genomic materials are capable of surviving, several tests are necessary. First, colonies must form and their ability to replicate efficiently measured. Hopefully, colonies would form, and replication would be efficient. Next, a Southern blot using a probe for the native genome should be performed. If possible, this same blot should be heated and washed to remove the original probe and a second probe specific for the transplanted genome should be hybridized. If a Southern blot cannot be manipulated as such, a second blot should be performed using this second probe specific for the transplanted genome. We would expect, if this experiment was successful, for no hybridization to be seen with the first probe. However, we would expect to see hybridization with the probe specific for the transplanted genome. Finally, the genome should be sequenced to determine what, if any, changes to the genome have occurred during these transformations. Hopefully minimal changes to the genome would occur during this process.
Southern blot 1
Probe->native genome
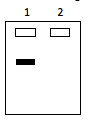
Southern blot 2
probe->transplanted genome
Lane 1 has host cells with native genome still present. Lane 2 has the genome of the transplanted cell. Both genomes are digested with the same restriction enzyme and DNA fragments are separated by gel electrophoresis.
It appears that the ultimate goal for this laboratory may be to completely synthesize a living cell. The lab has begun by synthesizing genomes using yeast and has now proven that transplantation of these genomes is possible. The next logical step is to prove that transplanted genomes can reproduce in cells that lack their own genetic material. Once this goal has been met, the lab may try to synthesize the cellular environment necessary for this synthesized genome from scratch. This is a very ambitious project with fruitful results likely far in the future. There is still a lot that we do not know about the cellular environment since it is such a dynamic system. Perhaps as we learn more about the cell, this project will become more feasible.