*This website was produced as an assignment for an undergratuate course at Davidson College.*
Creating Bacterial Strains from Genomes That Have been Cloned and Engineered in Yeast
Carole Lartigue, Sanjay Vashee, Mikkel A. Algire, Ray-Yuan Chuang, Gwynedd A. Benders, Li Ma, Vladimir N. Noskov, Evgeniya A. Denisova, Daniel G. Gibson, Nacyra Assad-Garcia, Nina Alperovich, David W. Thomas, Chuck Merryman, Clyde A. Hutchison III, Hamilton O. Smith, J. Craig Venter, John I. Glass
Science Vol 325 25 September 2009
Brief Overview:
The authors of this paper cloned an entire genome of a bacteria (Mycoplasma mycoides) in a yeast cell, genetically engineered it, then reinserted it into another species of bacteria, Mycoplasma capricolum. Thus, creating a new strain of Mycoplasma mycoides that had never existed. The Mycoplasma mycoides genome was modified by using the vast amount of methods that are readily available to take advantage of the yeast's machinery.
Results:
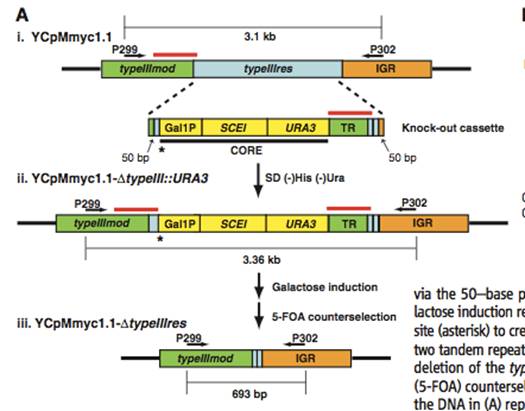
Figure 1A– This genetic map depicts a specific region, and details the step-by-step genetic modifications that were undergone to create a viable Mycoplasma cerevisiae genome. The vector that was used to introduce the bacterial genome into the yeast contained a selectable tetracycline-resistance marker, β-galactosidase gene, yeast auxotrophic marker, yeast centromere, and a yeast autonomously replicating sequence. YCpMmyc1.1 is the name of this genomic clone, and the yeast strain was a W303a. It was necessary for this process to occur in the yeast S. cerevisiae, because the modifications required could not occur with the “genetic tools available for the bacterium” (Lartigue et al., 2009). The black arrows above the DNA in Figure 1A (i, ii, and iii) represents PCR primers that were used to check for the appropriate changes. Figure1A (i) shows the deletion of the 3.1 kb non–required TypeIII restriction endonuclease gene (typeIIIres), which would normally code for an enzyme that cleaves DNA. To accomplish this, they created a knockout cassette containing a URA3 marker and a SCEI endonuclease gene that was under the control of a GAL1 promoter. Figure1A(ii)– When the yeast was transformed, the transformed genome was selected for by growth on a His (-) and Ura (-) medium. If the yeast was transformed, it contained a deleted typeIIIres gene, and contained the new 3.36 kb knockout cassette in its place. When the knockout cassette is induced to galactose, the Gal1 promoter activates the SCEI endonuclease gene, expressing the I-Sce I restriction enzyme, cleaving the knockout cassette where the asterik occurs, creating a break that promotes recombination between the two red lines-look to (iii). Figure 1A(iii)– This is the final desired product that has the typeIIIres gene, and the knockout cassette spliced out. This was seen after figure 1A(ii) has undergone galactose induction, and selection with 5-fluorootic acid to determine clones without the knockout cassette.
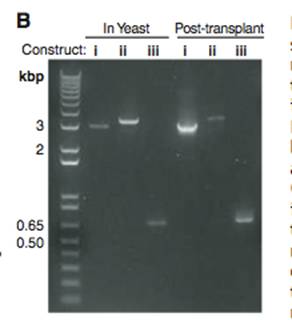
Figure 1B- This is a PCR gel that is used to check for the verification of the changes in the genome. In this figure, the constructs are referencing the previous figure. The PCR primers (black arrows in Figure1A(i, ii, and iii)) of the transformed genomes of Figure1A(i)-YCpMmyc1.1, Figure1A(ii)-YCpMmyc1.1ΔtypeIII::URA3, and Figure1A(iii)-YCpMmyc1.1ΔtypeIIIres were induced to PCR, and the gel was used to equate the bands to the three possibilities of transformed genomes. Construct i is slightly smaller than the band for construct ii, but much larger than the construct iii band, and it has a molecular weight of about 3, equating this lane with Figure1A(i)-YCpMmyc1.1. Construct ii is slightly larger than construct i, and this is because construct ii contains the knockout construct in place of the typeIIIres gene, equating it with Figure1A(ii)-YCpMmyc1.1ΔtypeIII::URA3. Construct iii has the smallest molecular weight, and this is because it contains neither the knockout cassette or the typeIIIres gene, equating this lane with Figure1A(iii)-YCpMmyc1.1ΔtypeIIIres. Additionally, the gel contains lanes after the post-transplant of the genome, and all of these bands have the same molecular weights as the constructs, indicating that the transplantation has occurred successfully.
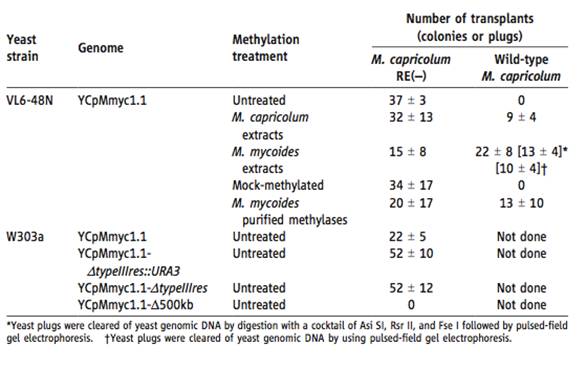
Table 1– This is a quantification of the results showing transplantion of the genomes. The results were determined by selecting for the growth of blue stained colonies, that grew on an SP4 medium with tetracycline at 37 degrees celcius. The number of transplants is an average of three or more experiments. Initially, no transplants were recovered for the YCpMmyc1.1 into the wild–type M. capricolum. As a result, it was deduced that a restriction enzyme in the M. capricolum degraded the unmethylated YCpMmyc1.1. As a result, two methods were undergone to try to fix the transplantation problem. First, the restriction enzyme of M. capricolum was inactivated. Secondly, the YCpMmcy1.1 of a VL6-48N yeast strain was methylated in vitro (not in a live organism, but a controlled environment) in a series of ways. Either by extracts from the M. capricolum, extracts from M. mycoides, mock-methylated, or M. mycoides purified methylases. The differently treated genomes were then introduced into the Wild-type M. capricolum, and the M. capricolum that contained the inactivated restriction enzyme (RE(-)). For the transplantation of the YCpMmyc1.1 into the M. capricolum RE(-), all methylated YCpMmyc1.1 resulted in colonies, and the number of colonies that formed were relatively similar, except for the M. mycoides extract methylation treatment, and the M. mycoides purified methylases, which contained the fewest amount. For the transplantations into the wild type M. capricolum, the mock-methylated and the unmethylated genome resulted in o colonies. Additionally, the table shows the transplantation results of the other YCpMmyc1.1 from the yeast strain W303a. None of these genomes were methylated. In this strain, the constructs created in Figure1A were used, as well as a large deletion clone (YCpMmyc1.1-Δ500kb) that lacked a 500kb sequence. This clone served as a control, because it was expected not work because the missing 500kb sequence contained essential genes. For this yeast strain, the genomes were only transplanted into the M. capricolum RE(-), and all forms of the genome resulted in colonies, however the YCpMmyc1.1 contained over 50% less colonies. As expected, the YCpMmyc1.1-Δ500kb genome resulted in no colonies. A key point for this table is that the bacterial genome can be cloned in both yeast strains.
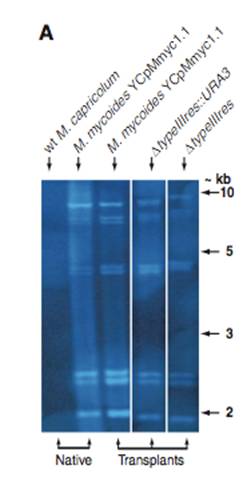
Figure 2A– This is a southern blot of M. mycoides and it was used to determine that the recovered colonies were indeed M. mycoides. For this blot, an M. mycoides specific probe was used, and the genomic DNA was induced to a Hind III restriction enzyme. The genomic DNA that was tested was native YCpMmyc1.1, transplanted YCpMmyc1.1 transplanted YCpMmyc1.1ΔtypeIII::URA3, transplanted YCpMmyc1.1ΔtypeIIIres, and wild type M. capricolum. Since, the probe is M. mycoides specific, a band is not expected to be seen in the wt M. capricolum lane, and it is not. Since bands are found in all other lanes, native and transplant, the recovered colonies were verified as M. mycoides. The wild type M. capricolum serves as a negative control, and the M. mycoides YCpMmyc1.1 serves as a positive control.
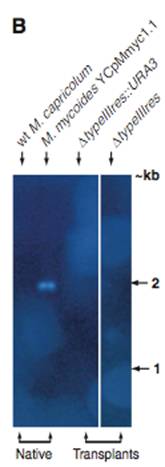
Figure 2B– A Southern blot that used the Type III restriction gene sequence as a probe. The genomes were restricted with Eco RV, and the tested genomes were the wild type M. capricolum, native M. mycoides YCpMmyc1.1, transplanted YCpMmyc1.1ΔtypeIII::URA3, and transplanted YCpMmyc1.1ΔtypeIIIres. Since, the type III restriction gene sequence was used, and this gene is only found in the native M. mycoides YCpMmyc1.1 a band should be present in this lane, and it is. The other transplants should do not have a band, because the Type III restriction gene was supposed to be deleted. No bands for these transplants are apparent, thus the Type III restriction gene was deleted. The wild type M. capricolum serves as a control.
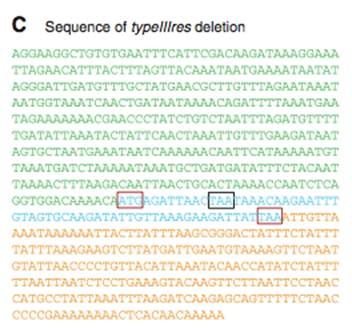
Figure 2C– A nucleotide sequence of the YCpMmyc1.1ΔtypeIIIres genome transplant. This shows that the Type III restriction gene was removed. The colors of the sequence match the genetic maps in Figure 1A. A small portion of the typeIIIres gene has remained, because of the overlap between the typeIIImod gene and typeIIIres gene (look to Fig 1A-red bars). The red boxes show the start and stop codons of the typeIIIres gene, and the black box shows the stop codon of the typeIIImod gene.
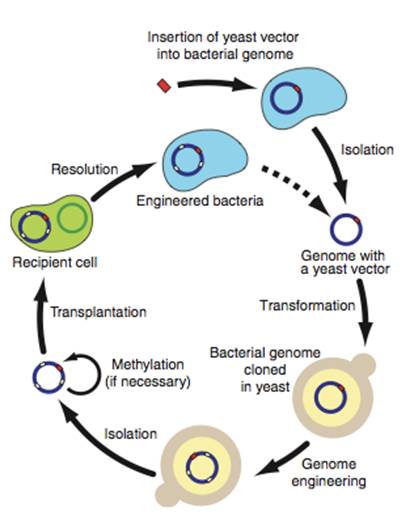
Figure 3– This is a recap/summarization of the process in which the bacterial genome was moved into a yeast, modified, and transplanted back into a bacterium. Through transformation, the yeast vector is inserted into a bacterial genome, then the new genome is cloned in the yeast. At this point, it is possible to create deletions, insertions, etc.The new genome is then isolated and transplanted into a recipient cell, creating an engineered bacterium. Additionally, after screening for a trait, this method can be redone to introduce the new bacterium to a new trait.
---------------------------------------------------------------------------------------------------------------------
Critique:
Overall, Lartigue et al., have done an amazing job with this work. The fact that they have been able to transfer a genome “between branches of life” is amazing (Lartigue et al., 2009). Additionally, they wrote in a manner that made the material very tangible. They did a good job of setting out to prove that the knockout cassette was actually deleted and that it was successfully transplanted into a new bacterium. They performed multiple, well-thought experiments to convince their reader that their methods can indeed create bacterial strains from cloned genomes in yeast. However, while their work was very good and extremely convincing, they made some small errors along the way.
First, I think it would have been nice to see pictures of the colony growth they quantified in Table 1. The material may be supplemented online, and while they do quantify their data, it would still be more convincing to see the actual colonies in the paper. Additionally, there is no evidence or thorough explanation of what happens to the recipient cell's DNA. How do they go about getting rid of it? Also, they mention that they sequenced the genome of one transplant, and that it matched M. mycoides, however, there is no evidence in the paper to show the exact match.
Table 1
It is odd that there is no negative control for the VL6-48N strain that is transplanted into the M. capricolum RE(-), but that there is one for the W303a strain. If one was present, it would be more helful. Why was the transplantation of the W303a yeast strain into the Wild-type M. capricolum not performed? Why were none of the W303a yeast strains methylated? Would they have the same number of colonies if they were? Fixing these problems would have created more solid results. Why does the W303a strain of the YCpMmyc1.1 genome have almost less than half as many colonies as the YCpMmyc1.1 genome for the VL6-48N strain when transplanted into a M. capricolum RE(-)? I understand that this is a methods paper, however it would have been nice to hear their explanation.
Figure 2A
Why was the southern blot for all the genomes not run on the same gel? The figure was pasted together for a total of 3 sections. It would have looked "neater" if it were all on the same gel. Additionally, it would have helped to see a loading control. This would help to determine why some bands may appear stronger than others.
Figure 2C
Why is there not a lane for the transplant of YCpMmyc1.1? Seeing this lane would have helped for a full comparison.
---------------------------------------------------------------------------------------------------------------------
Future Research
I think the first step will be to determine if this method will be successful in bacteria outside the realms of Mycoplasma? Could the genome be transplanted into another bacterium, like E. coli? To test this, the only thing that would need to be done is to apply the experimental methods described in this paper to another bacterium. It is also, as the authors say, possible to create deletions, insertions, etc. into the M. mycoides, thus creating mutated populations that can be studied. The methods presented in this paper will also allow other scientists to set out on creating a completely synthetic genome by following the cyclical process of genetic modification (Fig. 3). In clinical application, however, Lartigue, et. al bring up a good point for the use of their methods in creating a live vaccine for the mycoides group.
For the application of how such methods could affect our lives, the authors of this paper set other scientists onto a good path. They mention that their technology can possibly accelerate the construction of live vaccine strains. I think that this is definitely where this research will lead others. The authors mention that the mycoides group cause major diseases in animals that are ruminants. However, upon further research it appears that Mycoplasmas, in general, are a wide cause of disease in ruminants (which are grass chewing animals, that create cud in their first stomach). In Nigeria, the sero-prevalence of Mycoplasma in goats is 92%, and multiple forms of the Mycoplasma are found in pneumonic lungs of goats (Khusiluka, 2006). Animals infected with Mycoplasma have symptoms of arthritis, conjunctiva, and mastitis, and the Mycoplasma can be passed through an animals milk to humans(Kinde, 1994). In the U.S., diseases in humans that are associated with M. mycoides have a 90% rate of illness and/or fatality (Kinde, 1994). While the disease is primarily a livestock disease, it does spread to humans and there have been two outbreaks in the U.S (Kinde, 1994). As a result, there is an urgent need to develop a cure. If I were working on creating a new vaccine for this bacterium, here is what I would do: First, I would insert the yeast vector into the genome of the M. mycoides. Then, clone the genome in a yeast, after I take the bacteria, and insert/knockout a gene that makes the bacteria's lethality decrease significantly. Then, I would re-insert the new genome back into another bacterium of a similar species. It would then be necessary to introduce the modified bacteria to a ruminant, such as a goat, and observe the effects of the infection. If the goat develops symptoms, but is still in decent health, and quickly gets over the infection, the newly created bacteria may be possible to act as a live vaccine for humans and other livestock .
---------------------------------------------------------------------------------------------------------------------
References
1) Lartigue, C, Vashee, S, Algire, MA, Chuang, RY, Benders, GA, Ma, L, Noskov, VN, Denisova, EA, Gibson, DG, Assad-Garcia, N, Alperovich, N, Thomas, DW, Merryman, C, Hutchison, CA 3rd, Smith, HO, Venter, JC, Glass, JI. 2009 September. Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science; 325 (5948): 1693-1696. PubMed
2) Kinde, H, DaMassa, AJ, Wakenell, RP. 1994. Mycoplasma infection in a commercial goat dairy caused by Mycoplasma agalactiae and Mycoplasma mycoides; J Vet Diagn Invest 6:423-427.PDF
3) Kusiluka, L, Kambarage, D. CHAPTER 4: DISEASES CAUSED BY MYCOPLASMA. Diseases of Small Ruminants A Handbook. 2006. http://www.smallstock.info/research/reports/R5499/ch4-mycoplasma.htm.
If you have questions or comments feel free to E-mail: Huhayes@davidson.edu