*This website was produced as an assignment for an undergratuate course at Davidson College.*
Jonathan Holzwarth's Paper Review
Molecuar Biology 2010
Before you read further, I recomend you download the paper and print it out to follow along
you can find the pdf file here
Creating Bacterial Strains from Genomes That Have Been Cloned and Engineered in Yeast
Lartigue et al.
The purpose of this paper is to describe methods that can be used to produce genetically modified bacterium cells. They claim to have achieved this by cloning a Mycoplasma mycoides genome into S. cerevisiae by homologous recombination. Once in the yeast they engineered the M. mycoides genome in yeast by deleting a nonessential type III restriction endonuclease gene. Why is this so important? As of now, there is no molecular technology to conduct this kind of deletion in bacterium. Therefore, this paper implies that the scientists have found a way to engineer bacterium genomes using any method available to bacterium and/or yeast. This paper has huge implications, but does its data support its claims? Lets take a look at the data presented.
Figure 1
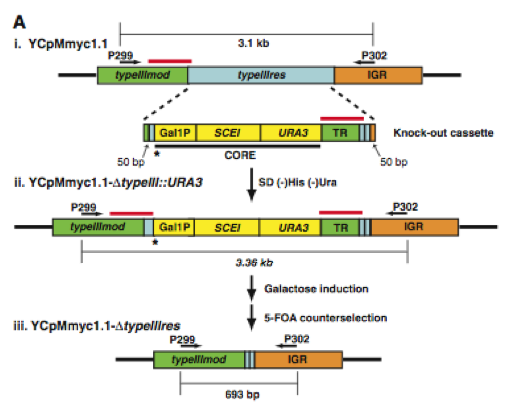
In the first figure, the authors provide two sections, A and B. Section A shows a simplified diagram of how they deleted the type III restriction endonuclease gene (typeIIIres). Section B shows a gel with PCR products from each step presented in section A.
Figure 1A. Here is a graphic of the procedure they used to delete the typeIIIres gene. (i) The top strand is the original M. mycoides genome (YCpMmyc1.1) that is now in yeast. Its important components are the two arrows depicting PCR primer sites for P299 and P302. The bottom strand is their "knock-out cassette" which is made up of homologous DNA (green, light blue, and orange), CORE DNA containing the GAL1 promoter controlling the SCEI endonuclease gene, and a URA3 marker (yellow), and tandem repeat DNA (green). The "knock-out cassette" was inserted into the yeast harboring the YCpMmyc1.1 strand. (ii) This strand is the stand selected in yeast by plating the cells on a His- and Ura- growth medium. The asterisk and the red lines above the DNA represent the two most important aspect of this strand. The asterisk shows the location of a galactose induced I-Sce I endonuclease site. When the endonuclease cleaves the DNA at this site, homologous recombination is promoted where the red line appear above the DNA strand. Recombination occurs and the deleted strands are selected using 5-flouroorotic acid (5-FOA) which selects against the Ura3 gene.
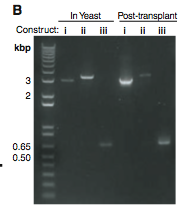
Figure 1B. This is a gel that shows the lengths of PCR products taken from yeast and M. capricolum (Post-transplant) cells. (i) These bands are PCR products of the original YCpMmyc1.1 DNA strand. Theses bands are shown to compare with the other two. They seem to be just about the predicted 3.1Kb. (ii) These bands represent the PCR product of the transplant clone with the knock-out cassette. They are definitely in the correct spot because about 100 base pairs were added when the knock-out cassette was added. (iii) These bands represent the PCR products of the transplant clone lacking the knock-out cassette. Once again they seem to be in the correct spot relative to the original DNA strand and the molecular weight marker.
Table 1
There are a lot of data available in this table. The first column distinguishes between two yeast strains they used to engineer and transplant the bacterial genome. The second column shows which genome was transplanted into the M. capricolum cells. The third column depicts the treatment given to the bacterium cells. The fourth and fifth columns show the number of cells with successful transplants. The target bacterium in the fourth column has a puromycin-resistance marker in the coding region of the single restriction enzyme (RE) in this strain of bacterium, whereas the target bacterium in the fifth column is wild type. The reason they used methylation treatments and created a M. capricolum RE- strain is to test thier hypothesis that a restriction enzyme degrades the unmethylated YCpMmyc1.1 DNA from the yeast. The researchers concluded that bacterial genomes could be stably cloned in two different yeast strains.
The blue arrows point out the important features of this table. The top arrow draws your attention to the fact that unmethylated wild type M. capricolum does not produce any viable transfers. This acts as a negative control because no transplants were recovered. Much like the first zero, the second zero is due to the fact that there are no transplants recovered because the restriction site is not blocked (mock methylated means the sample was treated the same as the methylated samples but not methyltransferases were added) and the enzyme gene is not silenced. These data along with the rest of the VL6-48N strain data suggest that it is necessary to inhibit the restriction pathway in M. capricolum in order to get successful transplantation of M. mycoide genome from yeast. The third arrow points to an insert from the yeast strain W303a. This strain was transplanted only in RE- M. capricolum and no transplant colonies were recovered. This data suggest that the bacteria genome with a large deletion (YCpMmyc1.1-500Kb) lacked essential genes but still had the bacteria YCp element. This clone served as a control because it shows the YCp element alone is not sufficient to cause transplantation back into bacterium calls. The asterisk and cross are also important feature of this table. They indicate that the yeast plugs (yeast genome) were taken out of the clones via digestion or pulse-field gel electrophoresis respectively. The fact that transplantation still occurred means that the yeast genomic DNA did not significantly alter the transplantation results.
Figure 2
There are three parts to figure 2. The first part is Southern blot analysis to verify the recovery of M. mycoides. The second part is also a Southern blot that shows the typeIIIres gene was in fact deleted. The third part is the sequence of the typeIIIres gene locus, and it provides more proof that the typeIII gene was, in fact, deleted from the genome.
Figure 2A. This figure is a Southern blot using M. mycoides-specific IS1296 element as a probe on Hind III-restricted genomic DNA to verify that M. mycoide was truly recovered. The first lane (left to right) is a negative control because it contains native wild type M. capricolum genomic DNA which should not bind to the probe. The second lane is the positive control because it is a native M. mycoides sample with the YCpMmyc1.1 genomic DNA. The next three lanes are samples obtained via transplantation. They all show the same band pattern as the native M. mycoides sample, providing strong evidence that the obtained transplantation samples are indeed M. mycoides.
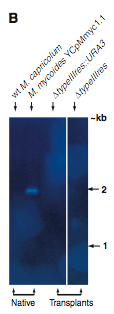
Figure 2B. This figure is also a Southern blot. However, this is probed with the typeIIIres gene sequence. The first two (reading left to right) lanes are negative and positive controls respectively. The native M.capticolum does not have the M. mycoides typeIIIres gene in its genome, and, of course, the M. myooices YCpMmyc1.1 has the typeIIIres gene in its genome. The next two lanes contain digested transplants, and no bands were detectable. This suggests that the transplants did indeed have their typreIIIres gene deleted.
Figure 2C. This is the sequence of the typeIIIres gene locus. The colors correlate to the colors in figure1A (i). The sequence matches the original M. mycoides YCpMmyc1.1. This data suggests that no recombination between yeast or recipient cell genomes with the donor genome (YCp). The red boxes depict the start and stop codons for the typeIIIres gene and the black box depicts the stop codon for the typeIIImod gene. The order of these codons indicates overlap between the two genes, and that overlap is why some of the typeIIIres gene remained after deletion.
Figure 3
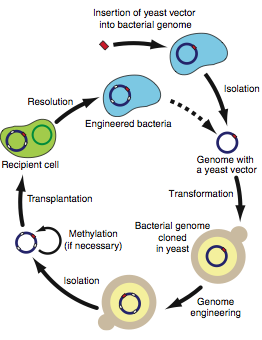
Figure 3 is a diagram depicting the protocol recommended for engineering bacterial genomes in yeast. It is pretty self-explanatorty. An important aspect of this diagram is the optional methylation step. You could either have transplantation target (recipient cell) that has its restriction enzyme genes silenced or you could methylate the important RE sites. The dashed arrow indicates the starting site repetition (newly engineered genome).
My Critique of the Figures and Table
Overall I thought the figures and table in this paper were satisfactory. However I would have added some information to improve upon them. For instance, the authors only mark on restriction site in the first figure. I would have liked to see where the Eco RV restriction site is on that strand of DNA. This would help improve upon Figure 2C as well because we see a 2 Kb band produced by digestion with the Eco RV site but its origin is not explained. The table had some aspects that made me skeptical as well. The first odd thing I noticed was that in the VL6-48N yeast strain, they did not try to transplant any of the YCp residues with the typeIIIres deletions. I am not sure if the just did not use this yeast strain to alter the bacterial genome, or if they just decided to focus on the original genome for testing their hypotheses about the restriction pathway. Either way it would have need more informative if they included data on the VL6-48N YCpMmyc1.1 with the knock-out cassette and the typeIIIres gene deletion. However, the lack of data in the top half of the table does not compare to the lack f data in the bottom half. In the W303a yeast strain, they tested the transplantation of all the YCp alterations. However, they did not test the transplantation rate with any methylation treatments so the only comparable data is the YCpMmyc1.1 transplant with no methylation treatment. They stated the "transplantation of all three YCp genomes into M. capricolum recipient cells resulted in similar numbers of tetracycline-resistant blue colonies." What I am wondering, and what the table they refer to does not explain, is how they compared all three YCp genome insertions to the VL6-48N counterpart when they only transplanted the native YCp genome using the VL6-48N yeast strain. Furthermore, the two data that are comparable do not show overlap in number of colonies (VL6-48N= 37+/-3, W303a=22+/-5). Another problem I have with the bottom half of the table is the fact that they did not try to transplant the genome into wild type M. capricolum targets. This leaves a taste of incompleteness and the extra data, if it showed similarities to the top half, would only support their claims. The fact that they did not do it means they did not have the time or resources, deemed it unnecessary, or it would hurt their claims. They never said why. Figure 2 is more compelling. The Southern blots show what they were looking for and the controls were selected well and worked well. The only problem is they do not tell us what to size band we should expect in figure 2B. Because their controls were so well thought out it seems the probe did, in fact bind to the typeIIIres gene. I also noticed they did not show the molecular weight (MW) marker and just put arrows a relevant points. I would prefer the MW marker with the arrows because their pictures of the exposed membrane seem to come from different gels. They may be aligned, but that does not mean the bands are al the same molecular weight. Figure 2C and Figure 3 are basic sequence and methods figures and they are explained by the authors well. I also believe their reasoning on why some of the typeIIIres gene remains in the deleted YCp genome based on their description of figure 2C. I agree with their claims that you can transfer bacterium genomes into yeast and engineer them. I also agree with the claim that they transplanted the engineered genomes back into different bacterium cells. I think their overall experiment was a success that leads to an innovative molecular method.
Design of Future Work
I have come up with two future experiments based on their discussion of this experiment:
Closing Comments and Reference
Overall, I believe this paper was extremely well organized and easy to understand. The implications of this method are huge, specifically the application to the study of bacterial strains similar to M. mycoide. This method may open a whole new door to the study of pathogenesis.
All figures and quotes come from the paper discussed:
Lartigue C. et al. "Creating Bacterial Strains from Genomes That Have Been Cloned and Engineered in Yeas." Science, 325. 25 September 2009. 1693-1696.
Please direct questions or comments about the webpage to Joholzwarth@davidson.edu
and questions and comments about the paper to svashee@jcvi.org