*This website was produced as an assignment for an undergratuate course at Davidson College.*
Review Paper: Whole Genome Rebooting
Brief Synopsis
In this paper, Lartigue et al describe a way to reboot the genome of a particular bacterium. They accomplish this goal by modifying the genome of bacterium #1 in yeast and inserting this modified genome into a bacterium #2 (which is a different species than bacterium #1), such that bacterium #2 takes on the “identity” of bacterium #1. In other words, these authors have found a way to synthesize, assemble and clone a bacterial genome in a eukaryotic organism and reinsert the re-engineered genome into a bacterium. This is beneficial because the genetic engineering that can be performed within a single bacterial cell is severly limited when compared to the genetic alternations that can take place within yeast cell. Thus, this technique opens up the possibilities to a large variety of genetic manipulations that can be performed on bacterial genomes that are not possible with the bacteria’s own machinery. This is very important because there many microbes that are important in industrial fields, such as pharmacy, that are very difficult to manipulate genetically (Lartigue et al., 2009). For example, biosynthesis of natural products by bacteria are very important components in the creation of novel drug therapies (Demain 2000). Being able to manipulate these organisms to produce an optimal level of a particular pharmaceutically important product could have great impacts not only in the pharmaceutical industry, but in medicine as well.
Key Points:
Bacterial genome that is modified in yeast (bacterium #1): Mycoplasma mycoides
Bacteria that the modified genome is inserted into and that takes on the “identity” of bacterium #1 (bacterium #2): Mycoplasma capricolum.
Summary of Results
Background
The first part of this experiment required the investigators to insert a yeast vector into M. mycoides. This was necessary so that when the bacterial genome of M. mycoides was inserted into yeast cells, the yeast cells would be able to function properly and manipulate the M. mycoides bacterial genome. The yeast vector contained a tetracycline resistance marker (to select for cells that had taken up the vector), the β-galactosidase gene (a visual screen for successful transformations), a yeast auxotrophic marker, a yeast ceontromere and a yeast autonomously replicating sequence (the last three allow for the proper replication of the bacterial genome in yeast). They were able to isolate one M. mycoides clone that had successfully integrated the yeast vector into the bacterial genome and they called this clone YCpMmyc1.1. They isolated this clone and transformed it into yeast cells. Figure 1 describes the mechanism by which the bacterial genome of M. mycoides is altered within the yeast cells.
Figure 1Ai: Shows the typeIIIres gene and the two genes that flank this gene in the YCpMmyc1.1 clone. The construct below the gene region shows the knockout cassette. The red lines above the gene region and the TR region of the knockout cassette indicate the tandem repeat sequences flanking typeIIIres that will allow for homologous recombination such that the knockout cassette will take the place of the typeIIIres gene in the YCpMmyc1.1 genome. P299 and P302 refer to the PCR primers that will bind to those particular regions in the genome in order to confirm that the deletion worked (Figure 1B). The CORE region of the knockout cassette is made up of Gal1P (Gal promoter), SCE1 (endonuclease) and URA3 (a marker for successful insertion of the knockout cassette).
Figure 1Aii: Shows what the typeIIIres gene locus looks like after homologous recombination with the knockout cassette. In order to select for clones that had successfully recombined this locus, the investigators grew the yeast in the absence of uracil, because the knockout cassette contains the uracil gene (ura3).
Figure 1Aiii: Once the cassette was inserted into the genome, the investigators induced expression of the Gal promoter with galactose, which allowed for the expression of Sce1 endonuclease. This enzyme cleaves the Sce1 site out of the knockout cassette. Deletion of this segement creates a break in dsDNA which promotes homologous recombination between the two tandem repeat sequences (red lines), which results in the deletion of the knockout cassette. Yeast clones were exposed to 5-fluoroorotic acid, which selects against the ura3 gene, so this counter selection makes sure that only the clones that have lost the knockout cassette are recovered.
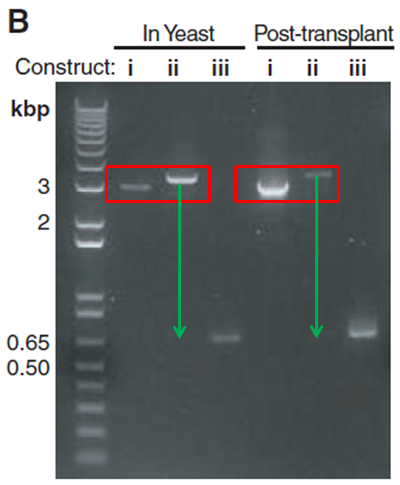
Figure 1B: This figure is a gel electrophoresis. This gel shows constructs isolated at the three different stages described in part A for both the YCpMmyc1.1 clone in yeast (“In Yeast”) and the transplant clones (“Post-transplant”). The “In Yeast” portion of the gel serves as a positive control to show what happens with the YCpMmyc1.1 genome in yeast cells. The “Post-transplant” portion of the gel shows the YCpMmyc1.1 genome that has been transplanted from yeast cells into M. capricolum cells. The gel shows that the banding pattern is very similar when performed with YCpMmyc1.1 in yeast and when performed with the YCpMmyc1.1 inserted into M. capricolum cells (Figure 1). In both cases, in between i and ii, there is a slight shift up which indicates insertion of the knockout cassette into the location where you had the typeIIIres gene (Figure 1). From ii to iii, there is a large shift down in molecular weight which shows that the knockout cassette has been deleted from the genome (Figure 1).
Image from Lartigue et al. 2009. Boxes and arrows not in original figure.
Figure 1B. This figure shows the verification PCR gel that these investigators ran in order to make sure that the knockout construct worked. The red boxes indicate the shift up in molecular weight after the insertion of the knockout cassette in the place of the typeIIIres gene. The green arrows indicate the shift down in molecular weight after the knockout cassette was deleted from the location of the typeIIIres gene. Notice that there is a slight discrepancy in the amount that the bands shift between the "In Yeast" and "Post-transplant" experiments.
Table 1
This table characterizes the effectiveness of transplanting the YCpMmyc1.1 genome that had been engineered (typeIIIres deletion) in yeast cells into M. capricolum bacterial cells. The investigators collected their data by counting the number of tetracycline resistant colonies that were blue (both the tetR and β-galactosidase genes were on the yeast vector). They found that in order to successfully transplant the YCpMmyc1.1 genome into M. capricolum cells, they had to inactivate the single restriction enzyme in M. capricolum (which they did by inserting a puromycin-resistance marker in the middle of the coding region for this enzyme), or methylate the donor DNA with either M. capricolum or M. mycoides extracts. Without the above two procedures, the inserted genome was digested by M. capricolum enzymes. In the VL6-48N yeast strain, untreated and mock-methylated YCpMmyc1.1 plasmids were not successfully transplanted into wild-type M. capricolum cells. However, all methylated, non-methylated and mock methylated YCpMmyc1.1 plasmids were successfully transplanted into M. capricolum cells that did not have the single restriction enzyme (M. capricolum RE(-)). In the W303a yeast strain, the investigators transplanted the YCpMmyc1.1 genome at different stages of the knockout process (shown in Figure 1A). They did not perform any methylation treatments for the DNA that was being transplated because they performed all of the transplantations in M. capricolum RE(-) cells. All of these transplants were successful except for the negative control where the investigators deleted 500 bp of the YCpMmyc1.1 genome such that essential genes were deleted.
Table 1. Transplantation of the M. mycoides genome that was engineered in yeast into M. capricolum cells. The red boxes highlight the idea that the absence of any methylation treatment resulted in zero successful transplants in wild-type M. capricolum.
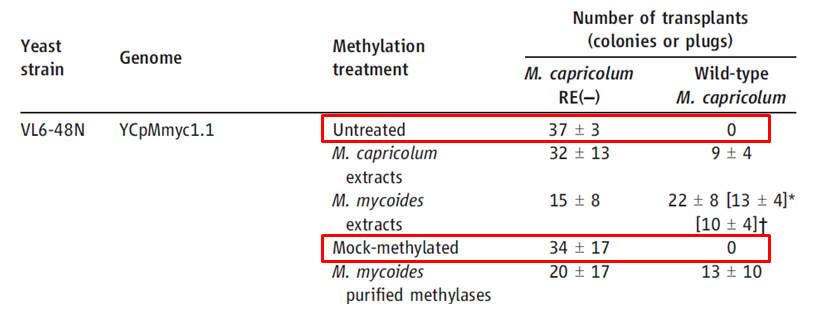
Table taken from Lartigue et al., 2009. Red boxes were not present in original figure
Figure 2A: This figure is a Southern Blot. The probe used in this experiment is specific for a genetic element found in the M. mycoides genome (IS1296).
Lane 1: Wild-type M. capricolum is probed with the M. mycoides specific probe. You should not see any bands, because the probe is specific for M. mycoides, so this lane serves as the negative control.
Lane 2: This lane serves as a positive control and shows the investigator where this probe binds to the YCpMmyc1.1 genome in yeast.
Lane 3: This lane shows where the probe binds when the YCpMmyc1.1 genome has been transplanted into M. capricolum cells. The banding pattern is the same as that seen in Lane 2. Thus, lane 3 serves to show that the transplanted version of theYCpMmyc1.1 genome remains unaltered when it is transplanted into M. capricolum cells as compared to this same genome in yeast.
Lane 4: This shows the banding pattern for the construct shown in Figure 1Aii. The top three bands have shifted up slightly. The bottom band has shifted down slightly. The overall banding pattern, however, is the same as is seen in the first three lanes.
Lane 5: This shows the banding pattern for the construct shown in Figure 1Aiii. Here again there seems to be a shift up in the top three bands and a shift down in the bottom most band, but the overall banding pattern remains the same.
*The fact that the overall banding pattern stays the same in the last three lanes (lanes 3-5) makes sense because the deletion they are creating is not in the region of the IS1296 genetic element, so this genetic element should stay intact throughout the engineering process.
Figure 2B. This figure is a Southern Blot. The probe for this blot was the typeIIIres gene sequence.
Lane 1: Wild-type M. capricolum is used as a negative control again. M. capricolum does not have a gene sequence complementary to the probe used in this experiment, so there is no band present in this lane.
Lane 2: The YCpMmyc1.1 genome is used as a positive control to show that the probe binds when the typeIIIres gene is present (shown in Figure 1Ai).
Lane 3: There is no band in this lane, which shows that the typeIIIres gene is absent when the knockout cassette is inserted into the region where this typeIIIres gene is located in the genome (shown in Figure 1Aii).
Lane 4: There is no band in the lane after the knockout cassette has been deleted from the typeIIIres gene locus (shown in Figure 1Aiii).
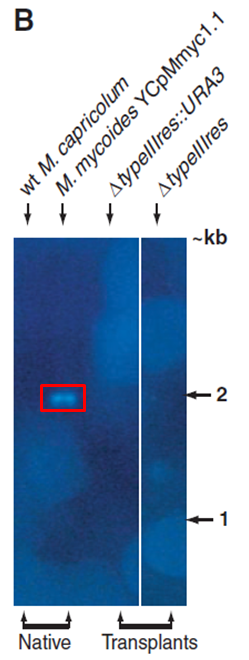
Image from Lartigue et al., 2009. Red box was not present in original figure.
Figure 2B. This figure shows the southern blot the investigators performed in order to verify that the typeIIIres gene had been deleted. They did this by probing the genomes indicated above each lane with the typeIIIres gene sequence. The red box indicates the only location that you should see a band which is in M. mycoides cells before the genomic engineering has taken place.
Figure 2C
This portion of the figure shows sequence analysis for the typeIIIres deletion. The seamless deletion in this region of the genome was varified by direct sequencing of the locus. The portion of the typeIIIres gene that remained after the gene was deleted is highlighted in blue. The green portion is typeIIImod gene and the orange portion is the IGR (intergenic region). The start and stop codons of the typeIIIres gene are boxed in red and the stop codon of the typeIIImod gene is boxed in black.
Figure 3
This diagram gives an overall schematic of the process that the investigators used in this paper. They show a way to transfer a genome between two brances of life: from bacteria, to a eukaryote and then back to bacteria. They first inserted a yeast vector (YCp) into a bacterial genome (M. mycoides).Then they isolated the bacterial genome that had incorporated the yeast vector (YCpMmyc1.1).They then transformed this cloned vector into yeast (two different strains: VL6-48N and W303a) and performed genome engineering with yeast genetic methods to create different alterations to the genome (in this case the typeIIIres gene deletion). The engineered genome was then isolated and inserted into the recipient cell (M. capricolum). Because the M. capricolum bacterium now has the genome of the M. mycoides bacterium, it has taken on the “identity” of M. mycoides. This process of genetic engineering can be used in a cyclical manner such that the engineered bacterium’s genome can be further manipulated (the dashed arrow) by further engineering (creating insertions, inversions etc…) the newly engineered genome.
Critique:
Overall, this paper provides a very convincing argument that it is possible to reboot the genome of an entire organism. They first give a detailed diagram of the knockout cassette and how it will function to create the engineered genome (in this case a deletion). They then show an experiment (gel electrophoresis after PCR) to convince the readers that this knockout cassette is functional in a living system. Next, they illustrate how the engineered genome will be inserted into another species of bacteria. This table is particularly compelling because they describe the problems of inserting a novel genome into a species and how they overcame these difficulties. The next figure serves to convince the reader that the transplanted version of the YCpMmyc1.1 genome has the same properties as the native YCpMmyc1.1 genome. They also provide good positive and negative controls in these experiment. Thus, the overall flow of information and the information found in each figure was compelling for the most part. However, there were some figures that could have been presented more effectively.
In Figure 1B, they do not provide any sort of explanation for why there is a difference in the brightness of the the different bands. They could have solved this problem by including a loading control, because there are multiple places on this gel where the bands are different intensities. The investigators also do not explain slight differences in band shifts between the two different sections of the gel ("In Yeast" vs. "Post-transplant"). These shifts in band size can be seen even more clearly with the annotation provided in Figure 1. In the "Post-transplant" portion of the gel, the band in lane "i" is much closer to the bottom of the red box than the same lane in the "In Yeast" portion of the gel (Figure 1). Additionally, the band in lane "ii" of the "Post-transplant" section appears closer to the top of the red box than the same lane in the "In Yeast" section of the gel, which makes the shift between lanes "ii" and "iii" in the "Post-transplant" section of the gel much larger than the same shift in the "In Yeast" section of the gel (Figure 1). Although these shifts do not appear to be significant, it would have been helpful for the investigators to address this issue. Because of these slight differences in size, I think it would have been especially important for these investigators to include a negative control lane where they ran a PCR with DNA that was not specific for the primers indicated, to make sure that there was no none specific binding of these primers to other regions of the genome. However, the lack of a negative control lane does not discredit their findings, because the fact that the overall banding pattern observed in both the "In Yeast" and "Post-transplant" experiments was very similar suggests that it would have been very unlikely if there had been non specific binding of the primers.
In Table 1, I felt that it would have been beneficial to include pictures of the plates where transplantations were successful versus plates where transplantations were not successful. Although the table was a clear way of summarizing the information, I think that including the pictures would have helped the reader better understand what it was that the investigators were looking at in order to come to the conclusions that they did.
In Figure 2A, lanes 4 and 5 are very difficult to interpret because the authors do not provide a restriction map to show where HindIII cuts this region (typeIIIres gene and the two genes flanking it) or were the IS1296 probe binds to the genome after it has been cut. Thus, it is very difficult to determine whether or not the banding pattern seen in the gel is what we should be seing. Thus, Figure 2C becomes very important, because this portion of the figure gives concrete evidence that the typeIIIres gene was actually deleted. I also thought that it was unnecessary for the blots in Figures 2A and 2B to be broken up because a single probe was used in Figure 2A and a single probe was used in fiugre 2B.Thus, it seems odd that they would need to cut up the blot as they have presented it in these two figures.
Future Experiments:
These investigators have discovered and characterized a very exciting new method for genetic engineering. Although they provide a very compelling argument for the importance and implications of this method, there are still many more experiments that can be done in order to improve and further characterize what these investigators have begun.
The first place I would start would be to attempt to engineer new modifications into the genome such as insertions and rearrangements. In order to make sure that these two types of modifications had in fact worked I would perform the following experiments for each kind of modification:
-Insertion:
Designing a construct for insertion: In order to do an insertion, I would use the idea of homologous recombination that these investigators used. However, instead of inserting a knockout cassette, I would insert the gene of interest. I would insert into a chromosomal location with a gene that was not necessary for the organism's survival (like the typeIIIres gene). I would also have an inducible promoter upstream of the gene I was interested in inserted. An inducible promoter would be more practical in this situation, because the native promoter for a particular gene may have other regulatory elements that would be very difficult to engineer into the genome with a single recombination event. I would also insert a selectable marker downstream of the gene being inserted that was flanked by unique restriction enzyme sites such that the selectable marker could be excised after I was able to select for colonies that had successfully recombined.
Detection: On a Southern Blot, I would use a probe that binds to sequence of the gene that is being inserted (Gene A in this example). I would also use a Western Blot analysis to determine if protein production has taken place. This would very important, because it is important to determine whether or not the protein can be properly produced in the newly engineered bacterium. If the insertion worked, then I would expect to see a band on the Southern Blot and I would expect there to be a band on the Western Blot as well.
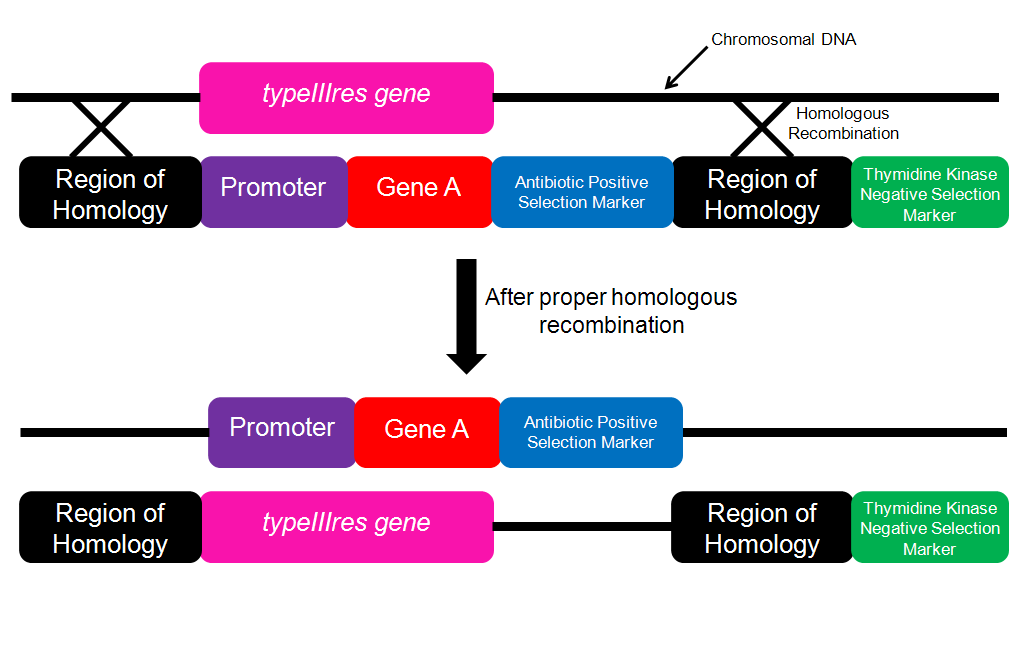
Figure 3. Schematic for performing gene insertion.The insertion cassette contains the gene of interest and an inducible promoter. If proper homologous recombination occurs, then the engineered genome could be probed with the Gene A sequence to determine whether or not the insertion was successful.
-Rearrangements:
Designing a construct for rearrangement: The concept of rearrangement could be acheived in mutliple ways. One manner in which rearrangement could be acheived would be by simply changing the order in which the genes appear in the genome. In order to acheive this, I would again use the idea of homologous recombination. On the construct that I introduced I would have the desired gene order (Figure 4). After proper homologous recombination (which would be determined by antibiotic selection for the presence of the antibiotic marker and the absence of the thymidine kinase gene), the genome would contain the desired gene order.
Detection: In order to detect whether or not homologous recombination had occured, I would cut the location where the rearrangement had occurred (I would isolate this region by finding two restriction enzymes that flank this site and then isolate this fragement by using a Southern Blot with a probe that bound to Gene A and Gene B to find the restriction fragment contained Gene A and Gene B) with the specific restriction enzyme that had its site in between Gene A and Gene B (Figure 4). If rearrangement had occurred, then I would expect to see two bands on the gel and if rearrangement had not occured, I would only see one band.
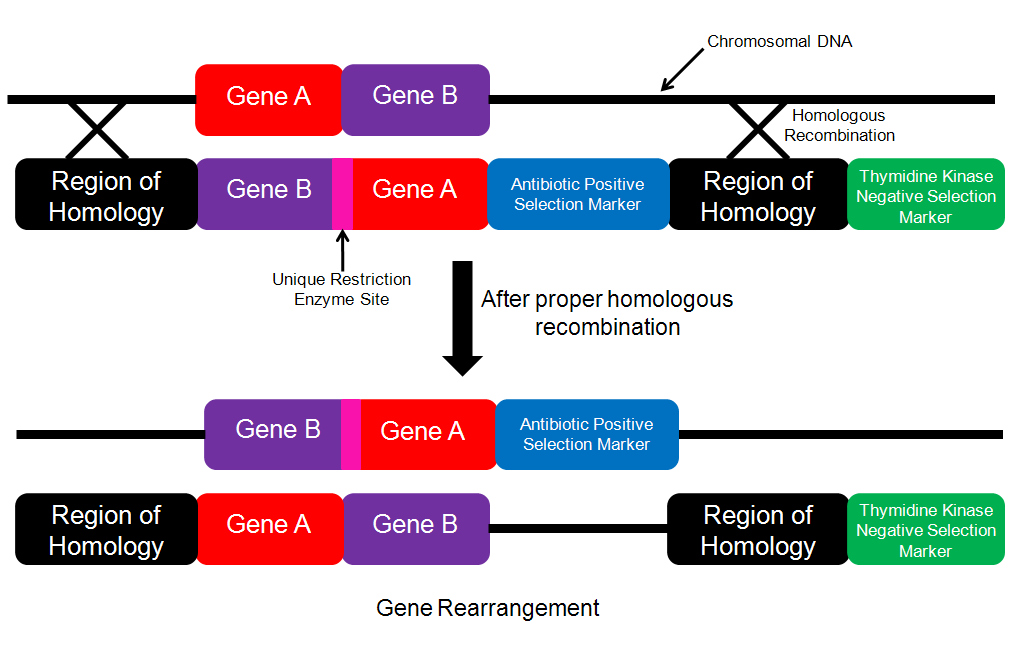
Figure 4. Schematic for performing gene rearrangement. The insertion cassette contains the genes in the desired order along with the positive selection marker within the region of homology. If proper homologous recombination has taken place, then the chromosomal DNA will be cut with the unique restriction enzyme.
I would then try the cyclical method of engineering that the investigators mention in Figure 3. I would perform the exact same set of experiments that the researchers described in this paper in order to genetically engineer a new element into the YCpMmyc1.1 genome. In order to test whether or not the engineering method had worked, I would use the tests described above. The authors also discuss engineering a combination of modifications in a single engineering event. I think that this would be a very important avenue to pursue, because it would seem that creating multiple modifications in a single engineering event would be important for creating engineered bacteria that will be useful in industry or medicine. The types of modifications that the investigators are trying to acheive would determine the experiments that they would need to do in order to determine whether or not the engineering event was successful or not. However, I would imagine that using a combination of the methods described in the paper and the methods I have suggested above would an appropriate manner in which to proceed.
I also think it would be interesting to look at whether or not it would be feasible to transplant an engineered genome into another genus. I think it would be particularly interesting to look at whether or not it would be feasible to transplant from E. coli into M. capricolum. E. coli are widely used bacteria in many fields, especially in the newly emerging field of synthetic biology. They are organisms that are very easy to manipulate and if this system were also feasible in E. coli, then that would open up a lot of possibilities in these field of synthetic biology. I would think that transplanting an engineered genome into a different genus would be feasible, but that there would be certain hurdles that would have to be overcome in transplantation phase like these investigators saw in the experiments associated with Table 1. I think that using the methods these investigators describe in Table 1 would be a good place to start to try and transplant a genome into a different genus.
References:
Demain AL. 2000. Small bugs, big business: the economic power of the microbe. Biotechnology Adavances 18: 499-514. http://www.ncbi.nlm.nih.gov/pubmed/14538099
Lartigue C, Vashee S, Algire MA, Chuang RY, Benders GA, Ma L, Noskov VN, Denisova EA, Gibson DG, Assad-Garcia N, Alperovich, Thomas DW, Merryman C, Hutchison CA, Smith HO, Venter JC and Glass JI. 2009. Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science 325: 1693-1696. http://www.ncbi.nlm.nih.gov/pubmed/19696314
Please direct questions or comments to Pallavi Penumetcha