This website was produced as an assignment for an undergratuate course at Davidson College.*
In this paper Lartigue et al. propose and attempt to verify a method for cloning a bacterial genome into another species (yeast). Basically the researchers propose that by taking a bacterial genome and integrating important yeast genes and selective markers, one can stably clone/transform such a genome into yeast. After cloning into yeast cells, the researchers suggest that alterations can be made to the bacterial genome using yeast genetic tools. Post-cloning, the researchers attempt to transplant the bacteria’s genome from yeast into a recipient bacterial cell that will then house the altered or unaltered bacterial genome from yeast. The overall steps are explained below along with an analysis of figures and a discussion on future research.
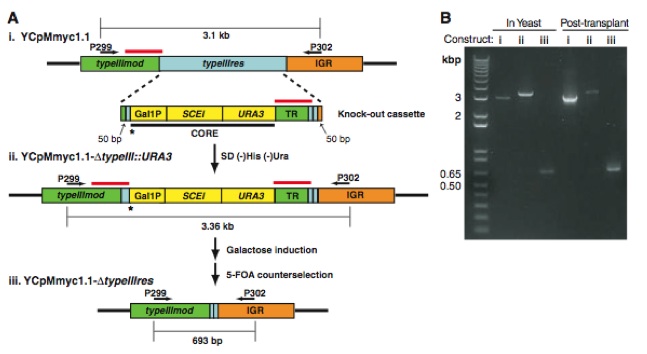
Figure 1: “Generation of Type II restriction enzyme deletions.”
(Lartigue et al., 2009)
The general purpose of the above figure is to explore and confirm the ability of researchers to change (engineer) the M. mycoides genome when it is in a yeast strain (which would mean that the researchers are actually engineering the YCpMmyc1.1 genome containing the M. mycoides genome). The researchers attempt to delete the nonessential Type III restriction enzyme gene (typelllres) from the YCpMmyc1.1 genome transformed in the yeast cells. They approach the deletion of the typelllres gene using a knock-out cassette (a fused sequence of DNA represented above). The knock-out cassette is meant to “knock-out” (meaning get rid of) the typelllres gene. The cassette is transformed into yeast cells, which harbor the YCpMmyc1.1 genome. The initial transformation of the cassette produced the YCpMmyc1.1- Δtypelll::URA3 genome, which no longer has the typelllres gene but does still contain the cassette. After inducing the Gal1 promoter in the cassette, the SCEI gene is activated and cleaves the cassette out of the genome leading to the final construct of YCpMmyc1.1-Δtypelllres (the construct that has the seamless deletion of the typelllres gene). The second part of figure 1 (part B) verifies that these changes to the YCpMmyc1.1 genome have occurred within yeast. In other words part B confirms whether or not the researchers are able to engineer bacterial genomes cloned within yeast. Part B is a simply gel on which PCR products are run. Note the arrows above each of the three constructs in part A to confirm where PCR primers (P299 and P302) for each construct begin and end. The figure shows appropriate band sizes (according to the MW marker in lane 1) for the in-yeast and post-transplant YCpMmyc1.1 constructs. The fact the band sizes are congruent with expected additions and deletions in the constructs while the YCpMmyc1.1 genome is within yeast or within M. capricolum after transplant, indicates that researchers have the ability to alter and engineer bacterial genomes while cloned within yeast cells using genetic tools only available for yeast.
While looking at this figure I like part A because I think the cassette mechanism used was a novel approach to deleting a specific gene. I also like that in part B you can see the intermediate step of the cassette insertion. Although the researchers tell us how long each construct is supposed to be, I would like to see the cassette run by itself in order to verify lengths. I also think the researchers could have done a better job explaining the Post-transplant columns on the gel, because although I think it is safe to assume that post-transplant refers to after genome transplantation of the YCpMmyc1.1 genome from yeast into M. capricolum, the researchers later bring up the problems with genome transplantation in figure 2 and that is not brought to light in this figure. Finally, I think it would have been a good idea to do the same engineering steps in the wt M. mycoides genome just to see the comparison or at least the YCpMmyc1.1 genome by itself, before transformed into yeast cells.
Table 1: “Transplantation of M. mycoides YCp genomes from yeast into wild-type and RE(-) M. capricolum recipient cells.”
(Lartigue et al., 2009)
The above figure explores the affect of proposed solutions to a problem the researchers encountered when trying to transplant the YCpMmyc1.1 genome from yeast into M. capricolum. While attempting to transplant the YCpMmyc1.1 genome from yeast (strain VL6-48N) into the wt M. capricolum cells, no transplantation occurred (which is indicated by a lack of colonies circled above in red). Researchers determined this lack of transplantation was due to the fact that the M. capricolum genome contained a gene that degraded the unmethylated YCpMmyc1.1 DNA. Researchers proposed two solutions:
1). Inactive the single gene in M. capricolum that degrades the YCpMmyc1.1 DNA by interrupting the gene sequence. The M. capricolum RE(-) represents the recipient cells that lack the gene which destroys YCpMmyc1.1 DNA.
2). Protect the YCpMmyc1.1 DNA by in vitro methylation of the YCpMmyc1.1 genome from the yeast strains using 4 different approaches: 1). Using extracts from M. capricolum, 2). Using extracts from M. mycoides, 3). Mock-methylation and 4). Using purified methylases from M. mycoides.
Table 1 summarizes the results from attempting both methods to force successful transplantation of the YCpMmyc1.1 genome from yeast (either strain VL6-48N or W303a) into M. capricolum. It is important to note that the number of colonies are the number of blue colonies and are therefore only the result of the YCpMmyc1.1 genome which contain the beta-galactose gene in the YCp vector. The two numbers circled in blue show successful transplanted into the M. capricolum RE(-) no matter what yeast strain the YCpMmyc1.1 genome was cloned in. These two results alone prove their first approach worked in correcting the original transplantation problem. The results in the purple box indicate that when the YCpMmyc1.1 genome from yeast VL6-48N is methylated in vitro by extracts from M. capricolum, extracts from M. mycoides, and purified methylases from M. mycoides, the DNA is protected in the wt M. capricolum. In other words, the researchers second approach to fix the original transplantation problem also works. The construct outlined in green is a deletion construct of the YCpMmyc1.1 genome from the yeast strain W303.a. In this construct essential M. mycoides genes were deleted and therefore there are no colonies in the M. capricolum RE(-). The fact that there are no colonies indicates that the transplantation was successful because after transplantation M. capricolum is now expressing the M. mycoides genes to live. This table basically notes that “the avoidance of the M. capricolum. . .” system that degrades the YCpMmyc1.1 DNA is essential to successful transplantation of the M. mycoides YCp genome (YCpMmyc1.1) cloned into yeast.
I think the use of the control (indicated in the green box) was a novel use of that deletion construct in this table. While reading the table, I contently asked myself why they felt it was necessary to apply both proposed “fix” techniques. I think it would have been interesting to show the techniques independently to see if one technique worked better (had more colonies). I also thought it was a weakness that they only did untreated YCpMmyc1.1 transplanted into wt M. capricolum with one yeast strain. It would have been nice to see this fact confirmed in both yeast strains because for all we know there is something in the yeast strain that may affect degradation. I also thought the “Not done” column should have been completed to provide further support for all constructs and that some in vitro methylation approaches should have been tested in yeast strain W303a. Overall, I think the table achieves what the researchers wanted to summarize- that their two proposed “fixes” to the original transplantation problem worked (at least in one yeast strain).
Figure 2: “Southern Blot of M. mycoides transplants”
The above figure 2 attempts to verify that the colonies recovered after transplantation are M. mycoides. When you transplant the M. mycoides YCp genome (YCpMmyc1.1) from yeast into M. capricolum, taking into account necessary methylation or using the M. capricolum RE(-) cells, the produced colonies should be phenotypically the same as M. mycoides (or at least YCpMmyc1.1). The first two images (A and B) are Southern Blots using a M. mycoides specific probe (ISI296) in part A and a probe for the typelllres gene that was deleted using the knock-out cassette in Figure 1.
Part A: The first two lanes show the controls for this experiment. The first lane should have no bands (which is true) because the probe is specific to M. mycoides. The second lane represents the M. mycoides YCpMmyc1.1 cells even before transformation into yeast. This lane shows you what band patterns should exist if the post-transplant cell contains the M. mycoides genome. The final three lanes in part A represent the different constructs of the YCpMmyc1.1 genome from yeast cells but after transplant into M. capricolum. The M. mycoides YCpMmyc1.1 from yeast does have any engineered deletions but as previously discussed the ?typelllres::URA3 and the ?typelllres are engineered constructs of the YCpMmyc1.1 genome in the yeast which have the knock-out cassette in place of the typelllres gene and a deleted typelllres gene respectively. As you can see the last three lanes look almost identical to the comparison lane 2. This indicates that after transplantation into the M. capricolum, the recipient cells contain the M. mycoides YCpMmyc1.1 genome. There does appear to be a slight band change in the last lane for the first two bands (as highlighted in yellow above), but I surmise that this is due to the deletion and possibly the deletion near a place where the probe binds (although logically you would believe that the deletion would create bands that run further).
Part B: The lanes in part B are almost identical to the lanes in part A (except for the YCpMmyc1.1 transplanted lane 3 in part A). Besides this one missing lane, the lanes represent the same things, except for this time the DNA is probed with the typelllres gene. In lane 1 there are no bands because typelllres does not exist in M. capricolum. Lane 2 shows a band for the typelllres gene, which makes sense because this genome is before transformation into yeast but more importantly has not been engineered so the typelllres gene is not deleted. In the final 2 lanes the typellres gene is missing, because in these two constructs the typelllres gene is either replaced by the knock-out cassette or it is seamlessly deleted. The fact that these two lanes represent genomes after transplantation indicates that you can engineer the YCpMmyc1.1 genome in yeast and transplant that engineered genome from yeast successfully (although this may be simply a result of a failed transplantation, because M. capricolum would also show no bars for typelllres).
Part C: This is a sequence analysis of the YCpMmyc1.1-?typelllres construct after transplantation into M. capricolum. This sequence analysis clears up the previous thought that transplantation may have just failed in the last two lanes of part B, because now we actually see that the sequences from those transplantions do align with M. mycoides and do contain the deleted segment. The colors correspond to the colors in Figure 1. The typelllres gene start and stop codon is boxed in red and the stop codon for typelllmod gene is in black. Although you can still see some of the typelllres sequence still exists after deletion, but this is because of the overlap between the typelllres gene and the typelllmod gene.
Overall this figure does a nice job of conveying the message that transplantations was successful, even when transplanting engineered YCpMmyc1.1 genomes from yeast. In part A and B it would have been useful to have MW markers run on the gel, especially because it appears as if the researchers used different gels. I would also suggest having part A on one whole gel and part B on one whole gel. In part B, I think the researchers should have included the engineered YCpMmyc1.1 native before transplantation to show that the typelllres probe did not bind there either. Figure 1 already suggested that changes in the engineered bacterial genome do not change whether or not the bacterial genome was cloned into yeast or was transplanted after cloning in yeast. I also think that it would have made a stronger case if they tested on the same gel in part B whether or not transplantation truly occurred for non-engineered strands. For this I would suggest still using the typelllres probe, but probe for the non-engineered strand after transplantation as well. The researchers also note that they tested to ensure that all post-transplantation genomes were entirely M. mycoides and not a mixture of M. mycoides and the yeast genome or the recipient cell genome (M. capricolum). I would prefer to view this data, which is not even referenced within the supplemental work.
Figure 3: “Moving a bacterial genome into yeast, engineering it, and installing it back into a bacterium by genome transplantation.”
Figure 3 represents the general methodology used to clone a bacterial genome in yeast and then transplant such a cloned and/or engineered bacterial genome from yeast into another species. These steps are outlined beneath the Overview section of this review, please reference that section for more detail.
Overall, I have no qualms with this figure. I like the coloring, specifically the use of coloring for the YCp vector. I do think that the resolution step could be clarified more.
The above figures suggest that one can take a bacterial genome, clone and engineer in yeast using genetic tools in yeast and then transplant the altered or copied bacterial genome back into a subspecies of the original bacterial genome. The researchers show in Figure 3 their general approach to moving, cloning and engineering a bacterial genome into yeast and then transplanting such a genome into recipient cells. Figure 1 shows the ability to engineer bacterial genomes in yeast and then transplant such engineered bacterial genomes (YCpMmyc1.1) into recipient cells (M. capricolum). Table 1 shows successful genomic transplantations of different YCpMmyc1.1 constructs from yeast into recipient cells. Finally Figure 2 verifies by Southern Blot analysis and sequencing that the genome transplanted during genomic transplantation of YCpMmyc1.1 (whether altered or unchanged) from yeast into M. capricolum, is the M. mycoides. Overall, I think this paper is extremely interesting. It talks about the ability to take a genome from one branch of life (prokaryote) and transfer/clone/engineer such a genome into another species of life (eukaryotic) and the ability to gain a new strain of M. mycoides through genome transplantation of the cloned and engineered bacterial genome in yeasts.
Stylistically I would suggest a stronger introduction to genomic transplantation. The researchers refer to previous work in their introduction and a brief synopsis of their past research would help with terminology. I thought that the proposed approaches to avoid the ability of M. capricolum to degrade donor DNA were both novel and I liked that the researchers used two different approaches and attempted to verify both in Table 1. The last figure was very strong and helped to understand the core steps.
The work of Lartigue et al. was groundbreaking. The ability to modify bacterial genomes within yeast cells extends the availability of yeast genetic tools to other species besides yeast. I would suggest a few experiments to further confirm the ability of engineered constructs to maintain throughout yeast cloning and genomic transplantation, such as making different changes besides a deletion to the YCpMmyc1.1 genome. In particular I suggest to study the effects when a particular gene is added as an alteration and whether or not such a gene could affect the host yeast strain and/or the product of genomic transplantation. I suggest using restriction enzyme sites or a transposon to study other affects of engineered YCpMmyc1.1. The same experimental tests could be used again to test the affects of other altered constructs. I also think it would be interesting to see if different yeast strains or “rescuing” methods to prevent degradation of YCpMmyc1.1 in recipient cells affected the ability to grow M. mycoides. I would also explore not only the Southern Blot data, but I think it would be interesting to study if any changes occur in the transcriptome (Western Blot) using transcriptome probes.
Not only are there improvements that can be made on the this paper in particular, but the introduction of this new method for utilizing yeast genetic tools to manipulate bacterial genomes in yeast opens up doors to explore engineering bacterial genomes in yeast. The paper suggests possible uses for this new method such as the ability to explore bacterial genomes (particularly M. mycoides) in a model system and possible the ability to speed up creation of vaccines because M. mycoides are causative agents f disease and can now be studied in yeast. With a new “tool-belt” of genetic tools in yeast, I suggest future studies into the regulation of multiple genes. For instance, one could alter multiple related genes within the bacterial genome (when it is cloned in yeast) using similar cassette methodology for deletions and possible insertions, along with transposons for insertions. If a person altered multiple genes in bacteria, one could study how those changes affected the bacterial genome once transplanted into a recipient cell. Specifically, one could study transcriptome output when multiple genes are altered or even how two specific genes interact or what pathway they belong to. I also think that this method shows promise for understanding how certain genomes evolve.
Lartigue et al. JI. 2009. “Creating Bacterial Strains from Genomes That Have Been Cloned and Engineered in Yeast.” Science [Internet]. [cited 2010 April 30]; 3 25:1693-1696. Available from: http://bio.davidson.edu/Courses/Molbio/restricted/2010/Genome_Reboot.pdf.
Please direct questions or comments to karicheson@davidson.edu