*This website was produced as an assignment for an undergraduate course at Davidson College.*
View Jmol structure of cholera toxin here.
Cholera toxin, responsible for the debilitating symptoms and effects of cholera infection, is secreted by the bacterium Vibrio cholerae. The first known cholera outbreaks began in the Indian subcontinent, and spread through trade routes via sailors and colonists to Russia, Western Europe, and North America. Cholera epidemics broke out in the 1800s, most notably in Russia, Germany, London, and Paris, claiming upwards of two million lives in one location. Cholera could strike and kill victims within four to six hours of infection, and victims often suffered a blue-black shriveled appearance due to the quick onset of severe dehydration and cramps. Cholera is no longer a significant threat in developed countries due to advances in public health measures, yet it still affects populations in Third World countries due to the lack of water filtration and chlorination.
Cholera toxin is a secreted heterohexameric AB5 enterotoxin (85 kDa) composed of enzymatic A and pentameric B subunits. It belongs to a class of microbial toxins which also have structurally independent enzymatic A and targeting B subunits (Spangler, 1992). Several other A-B toxins include Escherichia coli heat-labile enterotoxin, shiga toxin, pertusis toxin, and diphtheria toxin. The structure of cholera toxin closely resembles heat-labile enterotoxin from Escherichia coli, sharing 80% sequence homology (Zhang et al., 1995).
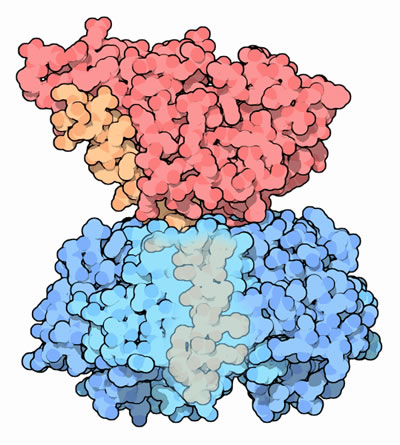
The wedge-shaped A1 subunit of cholera toxin sits above the ring of five identical B subunits, loosely held together by a connecting A2 chain (Figure 1). The B pentamer binds to GM1 gangliosides exposed on the luminal surface of intestinal epithelial cells (Sattler et al., 1978). The A subunit is activated by proteolytic cleavage and reduction of a single disulfide bond, which allows the free A1 subunit to pass through the cell membrane via endocytosis (Zhang et al., 1995). The enzymatic A subunit can then bind to NAD and permanently activate G-proteins, which in turn affects the secondary messenger system by causing sustained adenylate cyclase activity and constitutive cAMP production. This sustained increase of cAMP causes activation of intestinal sodium pumps, resulting in significant electrolyte and water loss, and sometimes life-threatening dehydration (Peterson & Ochoa, 1989).
Obtained from RCSB Protein Data Bank. Permission Pending
Figure 1. Cholera toxin. The A1 subunit (red) is held high above the B subunit (blue) with a tethering A2 chain (orange). Part of the B subunit is transparent to show the tail of the A2 chain.
The A and B subunits alone are not cytotoxic. While the enzymatic A subunit is capable of toxicity within the cell, it cannot function unless the B subunit gains entry into the cell first. The B pentamer is required for receptor binding and association with the target cell (Spangler 1992). Once the B pentamer has bound to the cell, the A subunit becomes activated as its internal disulfide bond (between A1 and A2 subunits) is cleaved. The A subunit is endocytosed into the cell; when it reaches the cytoplasm, it can no longer infect another cell and is instead sloughed off with that particular cell. Because the remaining extracellular B subunit is non-toxic, the cholera toxin is thus “self-limiting” in that cholera infection can only be propagated through the continual production and secretion of cholera toxin into the intestinal lumen (Spangler, 1992). The toxin will continue to accumulate in the intestinal lumen until it is purged out of the gut through the characteristic symptom of diarrhea.
Targeting B subunit
The structure of the targeting B subunit promotes detection and binding of GM1 gangliosides. The B subunits of cholera toxin assume a stable pentamer shape with five-fold symmetry (Figure 2). Each of the B monomers surround the central pore in the form of five amphipathic alpha-helices, which contribute to strong pentamer stabilization. The pentamer has five sites for GM1 ganglioside binding, one on each monomer, and it is believed that the cholera toxin binds to GM1 ganglioside with the B pentamer facing the membrane surface (Zhang et al., 1992). The presence of five separate GM1 ganglioside binding sites in one unit of toxin suggests that cholera toxin is highly efficient in recognition of target cells, which might explain how cholera toxin is able to affect its victims so quickly after infection.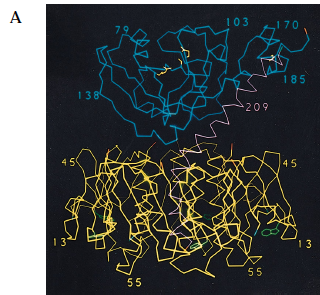
Obtained from Zhang et al., 1992. Permission pending.
Figure 2. Crystal structure of cholera toxin. (A) The A1 (blue) and A2 (magenta) subunits are held together by a single disulfide bond (yellow). The B (yellow) subunits assume a stable pentamer that surrounds the central pore. Each of the B monomers carry a GM1 ganglioside binding site (green). Notice that there is limited interaction between the A1 subunit and the B pentamer, and that the two are loosely held together through the tethering of the A2 chain. (B) The figure has been rotated 90 degrees to show the central pore in the middle, with the B pentamer surrounding it. Each of the GM1 ganglioside-binding sites (green) can be seen within the B subunits (yellow).
A subunit
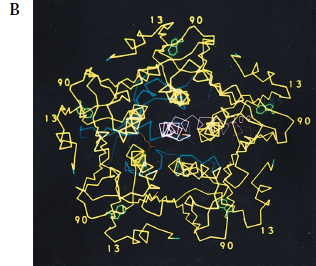
The A subunit is translated as one single protein (240 amino acids) that is nicked by a bacterial endoprotease to form the enzymatic A1 (residues 1-194) and connector A2 (residues 195-240) chains (Tomasi et al., 1979). Both subunits are loosely folded and characterized by a lack of secondary structure. The nicked A subunit is held together by non-covalent forces, specifically Van der Waals interactions, as well as a single disulfide bond between cysteine residues 187 and 199 (Figure 3, Zhang et al., 1992). The loose bonding pattern between the A1 and A2 chains seems to allow for a more rapid onset since only few interactions and forces must be overcome to release the toxic A1 subunit.
Obtained from Zhang et al., 1992. Permission pending.
Figure 3. A1-A2 subunits of cholera toxin. The catalytic A subunit is translated as one single protein that is eventually nicked by a bacterial endoprotease to form the A1 (magenta) and A2 (yellow) chains. The charge of the side chains of the amino acids are indicated by color: green for nonpolar, red for negatively charged, blue for positively charged.
Enzymatic A1 chain
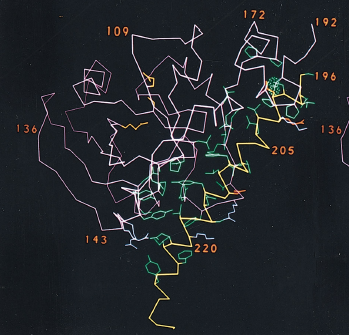
The A1 chain is the cytotoxic part of cholera toxin and is capable of binding NAD and catalyzing the ADP-ribosylation of G-protein (Spangler, 1992). The structure of the A1 chain is responsible for its toxicity and the subsequent excessive accumulation of salt and water in the intestinal lumen. The A1 chain is composed of three unique structures: A1-1, A1-2, and A1-3 (Figure 4). A1-1 (residues 1-132) consists of a globular unit that is a mixture of alpha-helices and beta-strands, and it is presumed that the NAD binding site sits within a remote cleft on this structure (Zhang et al., 1992). A1-2 substructure (residues 133-161) is an extended bridge between A1-1 and A1-3, and links the A1-1 substructure to the A1/A2 interface. This suggests that the second substructure has a similar function to the tethering A2 chain. It is important to note that A1-2 provides flexibility for the entire A1 chain. The third substructure is a globular unit that surrounds the disulfide bridge connecting A1 and A2 chains. It has a high density of hydrophobic amino acids (Zhang et al., 1992), which suggests a method for passing through the membrane bilayer during infection.
Tethering A2 chain
The A2 chain connects the A1 chain to the B pentamer. There are very few direct stabilizing interactions between the A1 chain and the B pentamer, which makes the A2 chain even more crucial towards the function of cholera toxin. The A2 subunit assumes a nearly complete alpha-helix structure except for the 52-degree kink in the central portion of the protein, which changes the direction of the helix for connection with the B subunit. The 52-degree kink is supported by a hydrogen bond between Ser228 and Asp229 (Zhang et al., 1992). The portion below the 52-degree kink sits inside the central pore of the B pentamer and intimately interacts with all five monomers of the B pentamer. The interactions between the A2 chain and B pentamer are mostly hydrophobic with several hydrogen bonds, which provide stabilizing support throughout the A2/B interface (Zhang et al., 1992). It was recently shown that mutated hydrophobic residues in the A2 chain and B subunit caused the A subunit to exhibit decreased stability and toxicity and increased susceptibility to proteolysis. In the same experiment, the B subunit was unable to form functional pentamers, which suggests that cholera toxin has a conserved hydrophobic region at the A2/B interface that is important for toxin assembly (Tinker et al., 2003).
The A2 chain shares a fairly extensive interface with the A1 chain – a disulfide bridge connects the two with multiple non-polar interactions running the length of the interface. Zhang et al. also found that the last four amino acid residues of the A2 chain (KDEL) mimic an endoplasmic retention signal. The continuous alpha-helix of the A2 chain allows the KDEL residues to sit protected within the central pore, suggesting that this endoplasmic retention signal has an important capacity for the toxin, possibly mediating internalization of the toxin into the cell (Rothmaan & Orci, 1992). When these terminal four residues were deleted from the protein, there was a significant reduction in the stability of the toxin (Streatfield et al., 1992), further indicating that the four residues may be necessary for successful functioning of the cholera toxin.
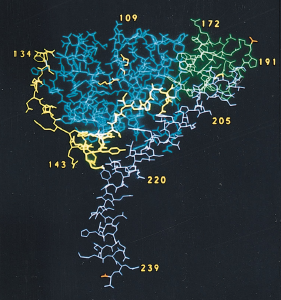
Obtained from Zhang et al., 1992. Permission pending.
Figure 4. Structure of A subunit. The A1 chain is composed of three different substructures: A1-1 globular unit (blue), A1-2 extended bridge (yellow), and A1-3 globular unit (green). The A2 chain (magenta) is composed of an almost-continuous alpha-helix chain, broken by one single kink in the central portion of the chain. The bottom portion of the A2 chain sits within the central pore of the B pentamer.
Peterson W and Ochoa L. 1989. Role of prostaglandins and cAMP in the secretory effects of cholera toxin. Science 245: 857-859.
Rothman J and Orci L. 1992. Molecular dissection of the secretary pathway. Nature 355: 409-415.
Sattler J, Schwarzmann G, Knack I, Rohm K and Wiegandt H. 1978. Studies of ligand binding to cholera toxin III, cooperativity of oligosaccharide binding. Hoppe-Seyler’s Z. Physiol. Chem. 359: 719-723.
Spangler B. 1992. Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiological Reviews 56: 622-647.
Streatfield S, Sandkvist M, Sixma T, Bagdasarian M, Hol W and Hirst T. 1992. Intermolecular interactions between the A-subunit and B-subunit of heat-labile enterotoxins from Escherichia coli promote holotoxin assembly and stability in vitro. Proceedings of the National Academy of Science 89: 12140-12144.
Tinker J, Erbe J, Hol W and Randall K. 2003. Cholera holotoxin assembly requires a hydrophobic domain at the AB5 interface: mutational analysis and development of an in vitro assembly system. Infection and Immunity 71: 4093-4101.
Tomasi M, Battistini A, Araco A, Roda L and D’Agnolo M. 1979. The role of the reactive disulfide bond in the interaction of cholera toxin functional regions. European Journal of Biochemistry 29: 8106-8111.
Zhang R, Scott D, Westbrook M, Nance S, Spangler B, Shipley G and Westbrook E. 1995. The three-dimensional crystal structure of cholera toxin. Journal of Molecular Biology 251: 563-573.
Please direct questions or comments to Teresa Wang