Excerpts taken from promotional material by Stratagene Cloning Systems
Timothy G. Simcox, Chao-Feng Zheng, Mary Ellen Simcox,
Peter Vaillancourt
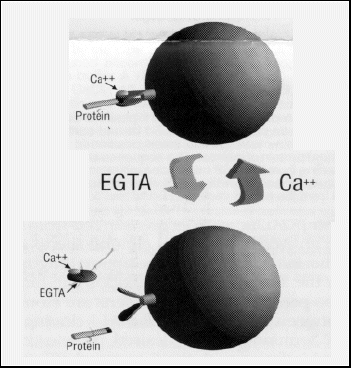
Stratagene has introduced a calmodulin-binding peptide (CBP) affinity-tag purification system that includes T7 RNA polymerase-based pET expression vectors containing the CBP affinity tag for N- or C-terminal fusion to recombinant proteins of interest. CBP fusion proteins are expressed at high levels in Escherichia coli and can be purified from crude cell extracts to near homogeneity with one pass through calmodulin affinity resin. The CBP tag binds to this resin with high affinity in the presence of low concentrations of calcium at neutral pH. Fusion proteins are eluted at neutral pH with 2 mM EGTA (Fig. 1). Therefore, both the binding and elusion of CBP fusion proteins are achieved using very moderate buffer conditions.
Figure 1. Calmodulin-Binding Peptide Affinity-Tag E.
coli Protein Purification System
In recent years, vector-based affinity-tag protein purification systems have become very popular. Initially, the only available systems used epitope fusion tags and antibody columns almost exclusively. These early systems produced low yields because of low-capacity antibody resin, and they often required harsh conditions or large, costly quantities of peptide to elute bound fusion protein. More recently, system improvements have provided higher-affinity interactions between immobilized receptor and ligand, affinity resins with greater binding capacity, and/or gentler binding and elusion conditions.
John Scott and coworkers at the Oregon Health Sciences University have developed an affinity-tag system for purifying proteins from bacteria. This CBP purification system1,2 is based on the relatively high affinity (Kd = 10-9) for the protein calmodulin exhibited by a 26-amino-acid C-terminal fragment from muscle myosin light-chain kinase, at physiological pH in the presence of calcium (figure 1).3 When calcium is removed from the environment, calmodulin undergoes a conformational change that results in the release of its ligand. The gentle binding and elusion characteristics make this system an excellent alternative to both the 6xHis and glutathione-S-transferase (GST) affinity-tag systems. The 4-kDa CBP affinity tag is significantly smaller than the 26-kDa GST tag so is less likely to affect the physical characteristics of the protein of interest.
Stratagene's newly introduced CBP Affinity-Tag Protein Expression and Purification System uses calmodulin affinity resin and a set of T7-based pET expression vectors that allow N- or C-terminal fusion of the recombinant protein of interest to the CBP affinity tag. The system also includes the site-specific protease thrombin for instances in which proteolytic removal of the affinity tag from purified protein is desired. We have also introduced a kit for the sensitive and rapid detection of CBP fusion proteins. Researchers also have the option of using cyclic AMP-dependent protein kinase (PKA) to radiolabel purified fusion proteins.
CBP Affinity-Tag Expression Vectors
To achieve tight control and high levels of induced expression of recombinant
protein, all three of the CBP affinity-tag vectors are derived from the
pET-11 series vectors (figure 2)4,5. These
vectors contain the T7/lacO promoter and a copy of the lacIq
gene to allow tight repression in strains bearing a source of T7 RNA
polymerase. Thus, in the absence of IPTG, the relatively high levels of
LacI protein expressed from these high-copy-number plasmids repress both
the expression of the lacUV5-driven T7 RNA polymerase and leaky expression
of cloned protein from the T7 promoter.
Stratagene's pCAL-n vector is derived from pET-11a and contains the CBP affinity tag followed by the 5-amino-acid thrombin-cleavage target positioned 5' to the multiple cloning site (MCS). DNA inserts containing protein-coding sequence are cloned such that hybrid proteins have the affinity tag fused in frame at the N terminus (figure 2A).
The pCAL-c and pCAL-kc vectors are derived from pET-11d and contain an Nco I N-terminal cloning site to allow positioning of the initiation codon for optimal high-level translation from the strong T7 gene10 ribosome-binding site, which is also present in pCAL-n. Inserts are cloned in these vectors between the Nco I and Bam H I cloning sites such that the C terminus of the desired protein-coding sequence is fused in frame with the C-terminal affinity tag located 3' to the Bam H I site. If the inserts contain one or more of the cloning sites, PCR primers may be engineered to include restriction sites that have overhangs compatible with Nco I (Afl III, BspH I or Sty I) or BamH I (Bgl II, Bcl I or BstY I). In pCAL-c, the thrombin-cleavage target is located immediately downstream of the Bam H I cloning site, followed by the CBP affinity-tag coding sequence. The pCAL-kc vector is identical to pCAL-c, except that the 9-amino-acid kemptide sequence is located between the Bam H I site and the thrombin-cleavage target. The kemptide is a synthetic PKA target that can be phosphorylated more efficiently than any of the known natural PKA target sites. The kemptide's presence on the fusion protein allows the purified protein to be labeled with 32P for use as a probe for interacting proteins.
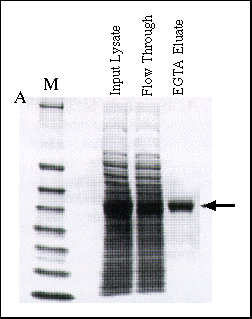
Figure 2 Affinity Purification of Fusion Proteins Tagged at C and
N Termini
Panel A: Purification of CBP-JNK. The protein CBP-JNK contains the affinity
tag at its N terminus. Epicurian Coli® BL21(DE3)pLysS competent
cells were transformed with the CBP-JNK expression plasmid, then a culture
was grown and induced according to standard protocol. The cell pellet from
a 20-ml culture was resuspended in 1.0 ml of lysis buffer (50 mM Tris-HCl
at pH 8.0, 150 mM NaCl, 10 mM ß-mercaptoethanol, 1 mM MgOAc, 1 mM
imidazole and 2 mM CaCl2) and lysed by sonication.
The cleared supernatent was incubated with 100 µl of calmodulin affinity
resin for 2 hours at 4°C on a rotating platform. The slurry was loaded
into a disposable column and washed with 3.0 ml of lysis buffer, then washed
again with 3.0 ml of lysis buffer containing 0.1 mM CaCl2.
Purified CBP-JNK was eluted with 5.0 ml of 50 mM Tris-HCl (pH 8.0) containing
150 mM NaCl 10 mM ß-mercaptoethanol and 2.0 mM EGTA. Fractions containing
CBP-JNK were concentrated, and equivalent amounts of the input lysate, flow-through
fraction and pooled, concentrated EGTA eluate fraction were electrophoresed
on a 4-20% SDS-PAGE gel and stained with Coomassie Brilliant Blue. The position
of the CBP-JNK fusion protein is indicated by the arrow.
CBP-Tagged Fusion Protein Affinity Purification
The protein CBP-JNK was derived by fusing the CBP-thrombin target affinity
tag to the N terminus of the ß-isoform of c-Jun N-terminal kinase.
Epicurian Coli® BL21(DE3)pLysS competent cells were transformed
with the CBP-JNK expression plasmid, then a culture was grown and induced
according to standard protocol.4 Lysates
were prepared in neutral pH buffer containing physiological salt and 2 mM
CaCl2, then batch incubated for 2 hours
at 4°C with calmodulin affinity resin (see figure 2 legend). The resin
was then applied to a disposable column, washed sequentially with 2 mM CaCl2 and 0.1 mM CaCl2-containing
buffers to remove unbound and nonspecifically bound proteins from the resin,
then eluted with 2 mM EGTA at neutral pH. As figure 2 shows, the
CBP-JNK fusion protein was present in high abundance in the input crude
lysate, is depleted in the calmodulin affinity resin flowthrough fraction
and is eluted to near homogeneity in the EGTA elusion fraction.

Figure 3 Proteolytic Removal of CBP Affinity Tag
Affinity-purified ß-Gal-CBP protein (490 ng), conatining the affinity
tag fused at its C terminus, was incubated with 0, 2, or 10 ng of thrombin
at room temperature for 2 hours and analyzed by SDS-PAGE. The arrows denote
full-length uncleaved fusion proteins and the mature cleavage products from
which the affinity tag has been enzymattically removed.
To allow removal of the CBP affinity tag following purification of the fusion protein, all three vectors encode short target sequences for the site-specific protease thrombin between the BamH I cloning sites and the CBP tag (or in the case of pCAL-kc, between the kemptide sequence and the CBP tag). Thrombin digestion of fusion proteins derived from the pCAL-n vector generates mature cleavage products that contain one exogenous amino acid from the thrombin target site (glycine) fused at the N terminus to the MCS. Cleavage of proteins derived from pCAL-c or pCAL-kc produces four amino acids fused at the C terminus to the BamH I cloning site or the kemptide, respectively. Figure 3 shows the results of thrombin digestion of proteins that had the CBP affinity tag fused at the C terminus of the protein. Figure 3 shows that, at a 1:50 ratio (10 ng of thrombin in this experiment), removal of the tag from the C terminus of a purified ß-galactosidase-CBP fusion protein expressed from the pCAL-c vector was complete after 2 hours.
Conclusions
Stratagene's CBP Affinity-Tag Protein Expression and Purification System
is a superior alternative to the currently available systems. The CBP tag
binds calmodulin with high affinity, yet binding and elusion conditions
are very gentle. In contrast, extremely high affinity tags such as polyhistidine
often require extreme conditions, such as low pH or high concentrations
of imidazole, to elute fusion protein from nickel-chelate resin.6 Unlike larger tags such as GST or the 40-kDa maltose-binding
protein, the 4-kDa CBP tag is less likely to affect the biological activity
or physical characteristics of the protein, so proteolytic removal of the
CBP tag is not needed as often. To date, as many as 30 proteins have been
purified using the CBP affinity tag,1, 2, 7, 8and
a resin capacity of 2 mg of CBP fusion protein per milliliter of resin is
achieved.1, 8
In addition to the above advantages, the availability of multiple affinity-tag purification and detection systems allows the researcher to study protein-protein interactions in vitro. The importance of the role of protein-protein interactions in mediating intracellular signal transduction has been well documented and is the object of intense study.9 Such interactions are being identified rapidly using the yeast two-hybrid system10, 11 and interaction cloning.12, 13 Characterization and validation of such interactions typically require in vitro binding studies. Because each component of an interacting pair can now be generated with a separate affinity tag in E. coli, the researcher can easily verify the direct interaction of proteins identified by genetic screens. Such experiments require the use of two or more different affinity tags. The mild binding and elusion conditions used with Stratagene's CBP Affinity-Tag Protein Expression and Purification System make it ideal for generating binding sites for these experiments. Furthermore, while the application of GST fusion proteins is limited by GST-mediated multimerization, the CBP affinity tag's lack of dimelization elements should allow accurate analysis of real-time kinetics and direct binding using the Pharmacia BIAcoreTM Biosensor system.
Acknowledgments
The authors would like to thank John Scott, Dan Carr, Anthony Means,
Tony Ribar, Charles Mena and Carolyn Erickson.
References
I. Stofko-Hahn, RE., Canr, D. W., and Scott, J. D. (1992) FEBS Lett.
302:274 278.
2. Carr, D. W., et al. (1991) J Biol Chem. 266: 14188-14192.
3. Means, A. R., et al. (1991) In Cellular Calcium, A Practical Approach
(J.G. McCormack and P.H. Cobbold, eds.) IRL Press Oxford.
4. Studier, F. W., et al. (1990) Methods Enzymol 185: 60-69.
5. Weiner, M. P., Anderson, C., Jerpseth, B Wells, S., Johnson-Browne, B.
and Vaillancourt, P. (1994) Strategies7: 41-43.
6. Schmitt, J., Hess, H., and Stunnenberg, H. G. (1993) Mol. Biol. Reports
18: 223-230.
7. Scott, J. Personal communication.
8. Simcox, T. G., Zheng, C F., Simcox, M E., and Vaillancourt, P. Unpublished
data.
9. Cohen, G. B., et al. (1995) Cell 80: 237-248.
10. Vojtek, A. S., Hollenberg, S. M., and Cooper, J. A. (1993) Cell 74:
205-214.
11 Mullinax, R. L., and Sorge, J A. ( 199S) Strategies 8: 3-5.
12. Guarente, D (1993) Proc. Natl Acad. Sci. USA 90: 1639-1641.
13. Skolnik, E. Y., et al. (1991) Cell 65 :83-90.
14. Lowenstein, E. J. (1992) Cell 70: 431-442.
Return to Molecular Biology Main Page
![]()
© Copyright
2000 Department of Biology, Davidson College, Davidson, NC 28036
Send comments, questions, and suggestions to: macampbell@davidson.edu