Summary:
This paper introduces a new
way to look at the evolutionary conservation of protein interactomes by
studying
the multiprotein complexes of a number of highly evolutionarily
divergent species.
The authors’ stated goal was to identify protein complexes and physical
protein-protein
interactions (PPIs) that are evolutionarily conserved across a wide
range of
phyla. They also studied the functional consequences of these PPIs. The
authors
analyzed protein complexes from five organisms: worms (Caenorhabditis elegans), fruit flies (Drosophila melanogaster), mice (Mus
musculus), sea urchins (Strongylocentrotus
purpuratus), and humans. They independently validated these data
using
analyses of frog (Xenopus laevis),
sea
anemone (Nematostella vectensis),
amoeba
(Dictyostelium discoideum),
and yeast (Saccharomyces
cerevisiae).
Evaluation:
I
was extremely impressed by
the amount of work that went into this paper. It was more of a basic
science
paper than translational research, but as a scientist it was
fascinating to see
how the authors were able to detect multiprotein complexes and their
evolutionary conservation. Additionally, the last figure showed that
many of
their predictions were functionally relevant. Therefore, this method
could have
important implications in nearly every field of biology since these
PPIs are
key to the function of cells and organisms. However, the authors
didn’t
explicitly state any of these implications. Rather, they left the
reader to
think of them. Therefore, I think it would have been helpful if they
had, for example,
further interpreted the importance of the results of Figure 5 to show
their
applications. Additionally, some of the figures were slightly
confusing and the
figure legends were not always very helpful in explaining them. My
main issues
were with the workflow in Figure 1, the protein complexes in Figure 3
(it was
difficult to see how the different parts of the figure related to each
other
and this was not explicitly stated in the legend), and the way they
presented
their AP/MS data in Figure 4a.
Figures:
Figure 1:
This figure shows the sequence of steps the authors
went through to obtain their results. First, the figure shows the
phylogenetic
relationships between the organisms studied (panel A). Next, the authors
display
sample data from the co-fractionation experiments that separated protein
complexes based on their biochemical properties as well as the MS/MS
experiments that were used to identify the proteins based on their mass
spectrometry
profiles (panel B). The right-hand side of panel B also shows the
outputs from
these analyses. Finally, the authors show the computational analyses
they
performed to identify conserved complexes (panel C). Proteins that
co-eluted
(suggesting that they interacted in
vivo)
were scored computationally to determine which interactions were
conserved in
at least two species. The program was trained based on known conserved
protein
complexes and was able to successfully identify other known protein
complexes
as well as novel complexes.
key
takeaways:
The authors laid out their
method and showed that they were able to successfully train their
program to
identify conserved protein complexes across five evolutionarily
disparate
species.
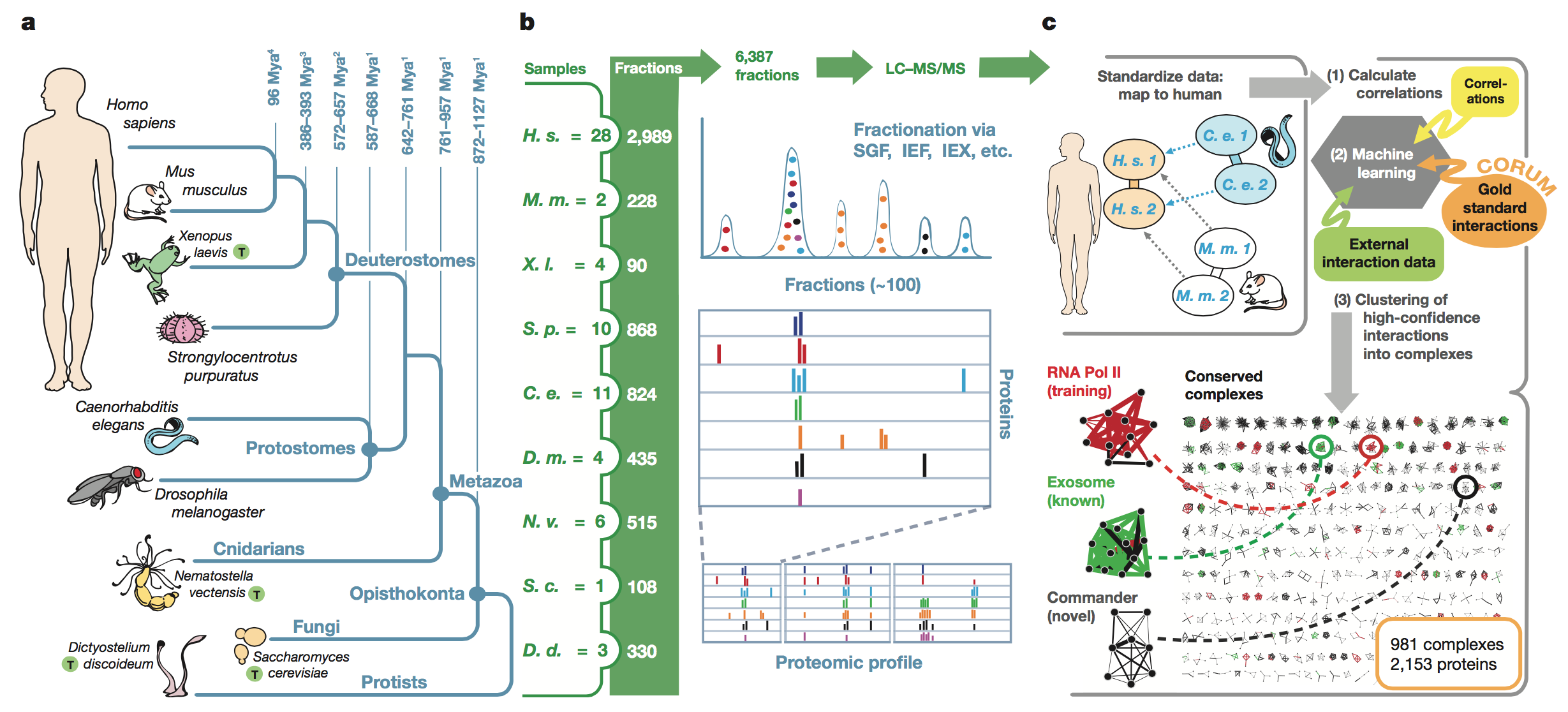
Figure 2: In
this figure, the authors use their computational
method to analyze protein co-complex interactions across the different
taxa
studied. They first show how they have expanded on their previous work
in the
current study by including new taxa and more human proteins (panel A).
They
also validate their method by demonstrating that they are able to use
their
co-fractionation experiments plus external evidence to more accurately
identify
co-fractionated proteins compared to either method alone (panel B). They
measure accuracy based on precision (the fraction of results that were
relevant)
and recall (the fraction of relevant instances retrieved). They also
show the
success of their method by confirming that complexes with high
co-elution
scores were predicted to interact based on their algorithm.
The authors further
validated their method by showing they were able to successfully detect
distinct subunits of a known protein across three species (panel C). The
protein they chose was the 30S subunit of the proteasome, which is a
complex
shaped like a tube with a lid on top. Co-fractionation data, a
correlation
matrix, and a hierarchical clustering dendrogram were all consistent
with the
known shape of the proteasome. Finally, the authors broke down the
conserved
co-complex interactions discovered to state whether they had previously
been
identified in other databases. They found that the majority of the PPIs
they
found were in fact novel.

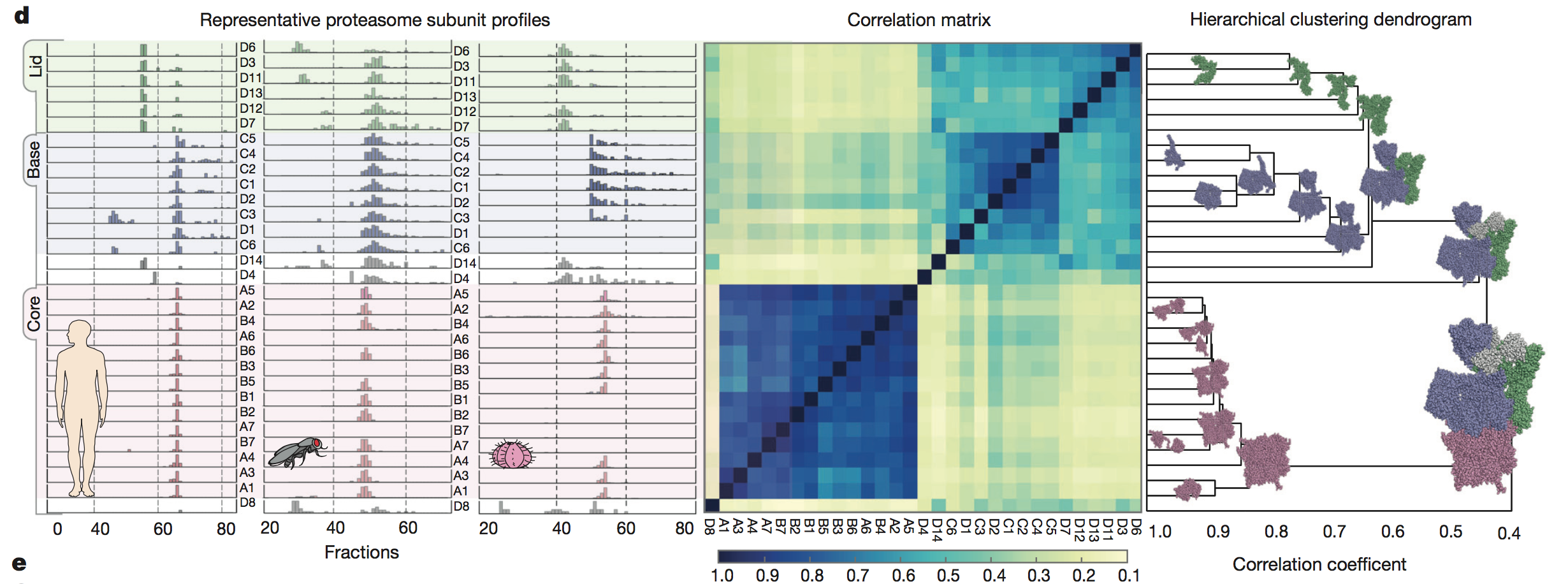
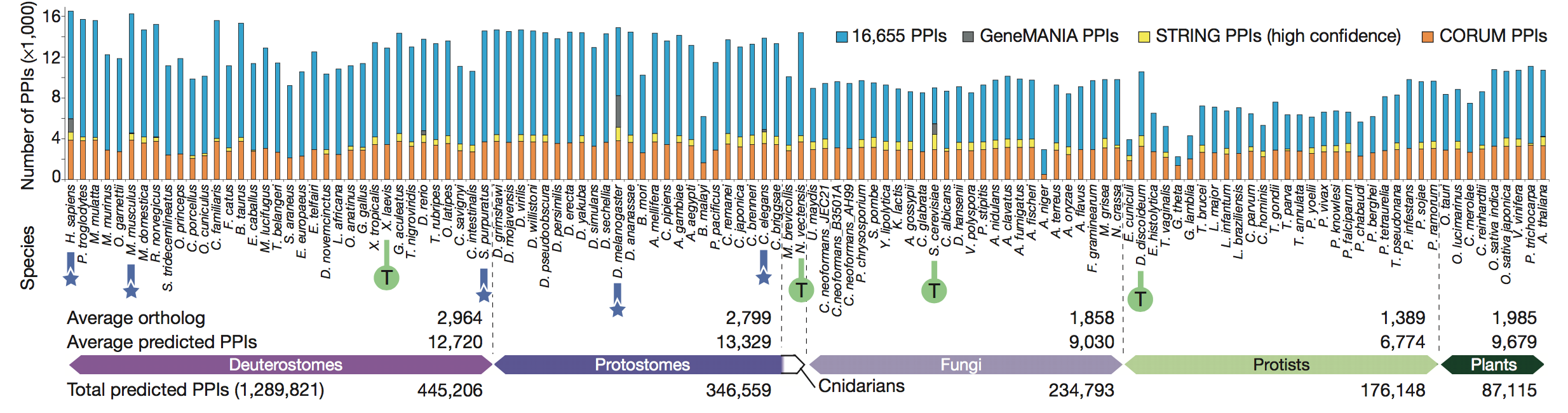
Figure 3: In
this figure, the authors looked at the conservation
of the 981 multiprotein complexes identified by their method, as well as
the
origins of the subunits of these complexes. They identified the subunits
as
being either “old,” meaning the complexes had component ages of over one
billion
years of evolutionary divergence, or “new,” meaning the complexes were
metazoan-specific (or only found only in animals) and had diverged
evolutionarily approximately 500 million years ago (panel A). Many
complexes
were made of mixed components, meaning they contained subunits that were
both
old and new. This figure also shows “zoomed-in” versions of six example
complexes that were either completely old, completely new, or mixed
(panel B).
Finally, the authors
showed the prevalence of the putative complexes across
phyla by looking at the conservation of the six complexes from panel B
across
phyla (panel C). They saw that all of the complexes tended to have
orthologs in
animals (Cnidarians, Protostomes, and Deuterostomes). On the other hand,
fewer
complexes containing new subunits had orthologs in fungi, protists, and
plants,
as seen by the dark blue areas (indicating lack of orthlogs) for
complexes 3-6 (which
contained new subunits) in phyla that were more evolutionarily distant
from
Metazoa.
key
takeaways: The rise of Metazoa, or
animals, led to the development of many new genes that encoded for
protein
subunits involved in multicellularity. However, “older” protein subunits
and
complexes are still conserved across phyla and are generally involved in
core
cellular processes.
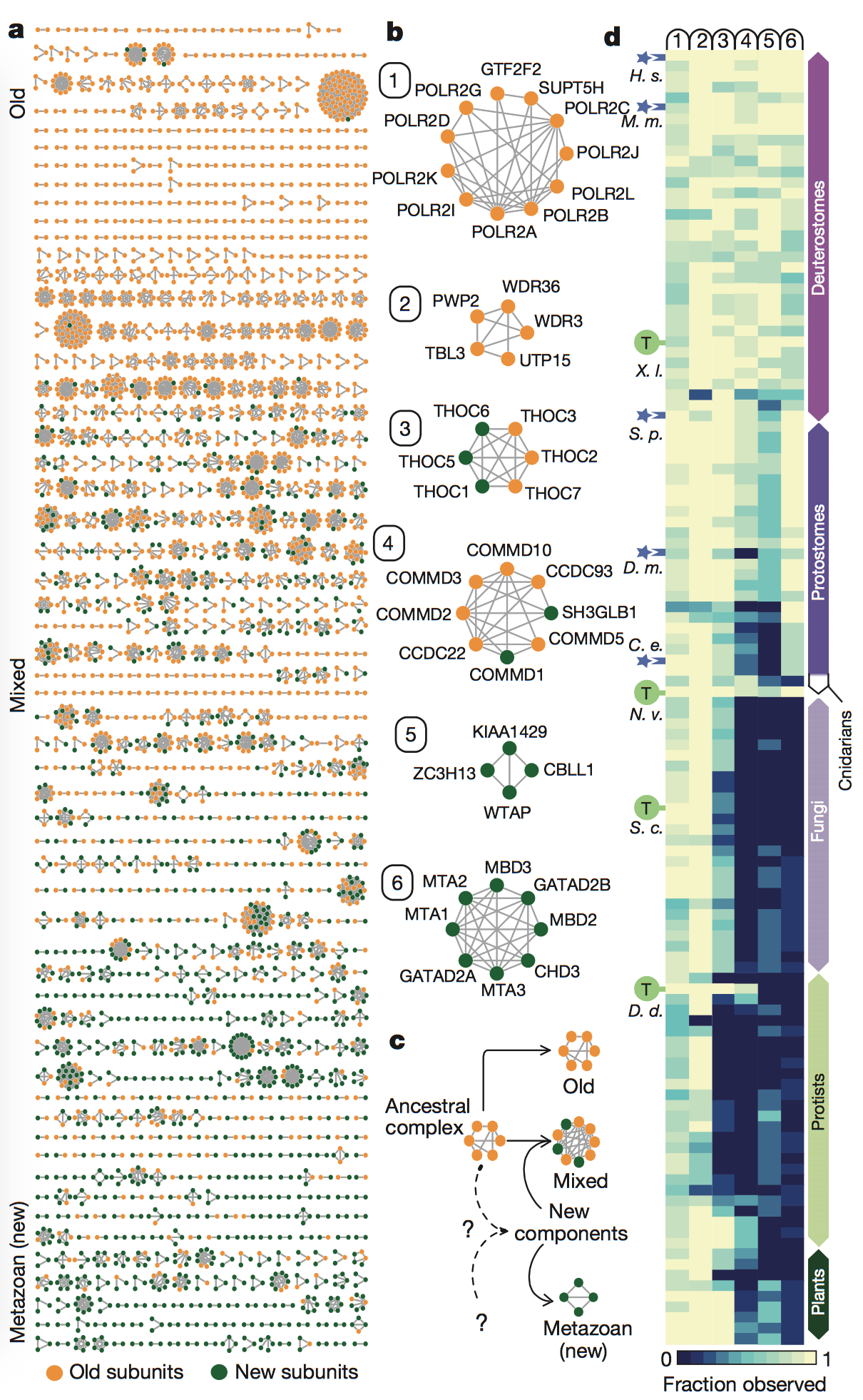
Figure 4:
Here, the authors attempted to verify the accuracy
of their predictions via physically analyzing the complexes they
identified
computationally. They performed affinity purification mass spectrometry
(AP/MS)
to verify the interactions among a number of their predicted complexes.
This
method uses one protein in the complex as “bait” to capture the other
proteins
by allowing them to bind to the bait. The proteins are then identified
via MS. The
results of these experiments showed that the predicted interacting
proteins
were almost always detected by human AP/MS, because the bait protein
(the
header in each box of the inset) caught its predicted interacting
proteins in
the vast majority of AP/MS trials (panel A).
The authors then
used data from
other papers to validate their results. They saw that the complexes they
predicted to be conserved overlapped with another AP/MS experiment
(panel B),
and also that the inferred MW of the human protein complexes
corresponded to
size-exclusion chromatography profiles from other studies both across a
wide
range of sizes (panel C) and for one specific complex whose subunits
were
co-eluted (panel D).
key
takeaways:
The authors confirmed the
accuracy of their predictions based on the physical properties of sample
protein complexes that they had identified using their method.
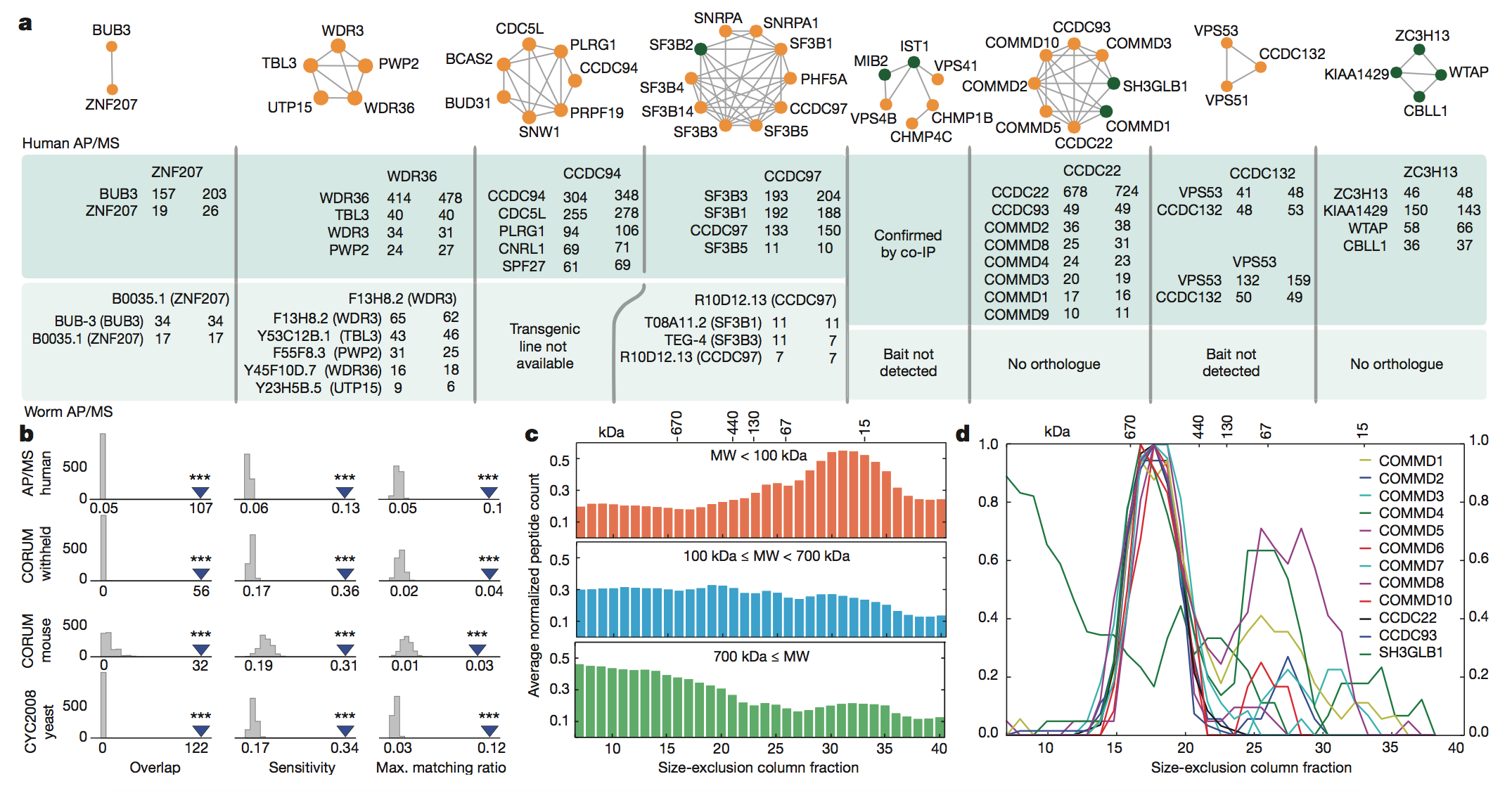
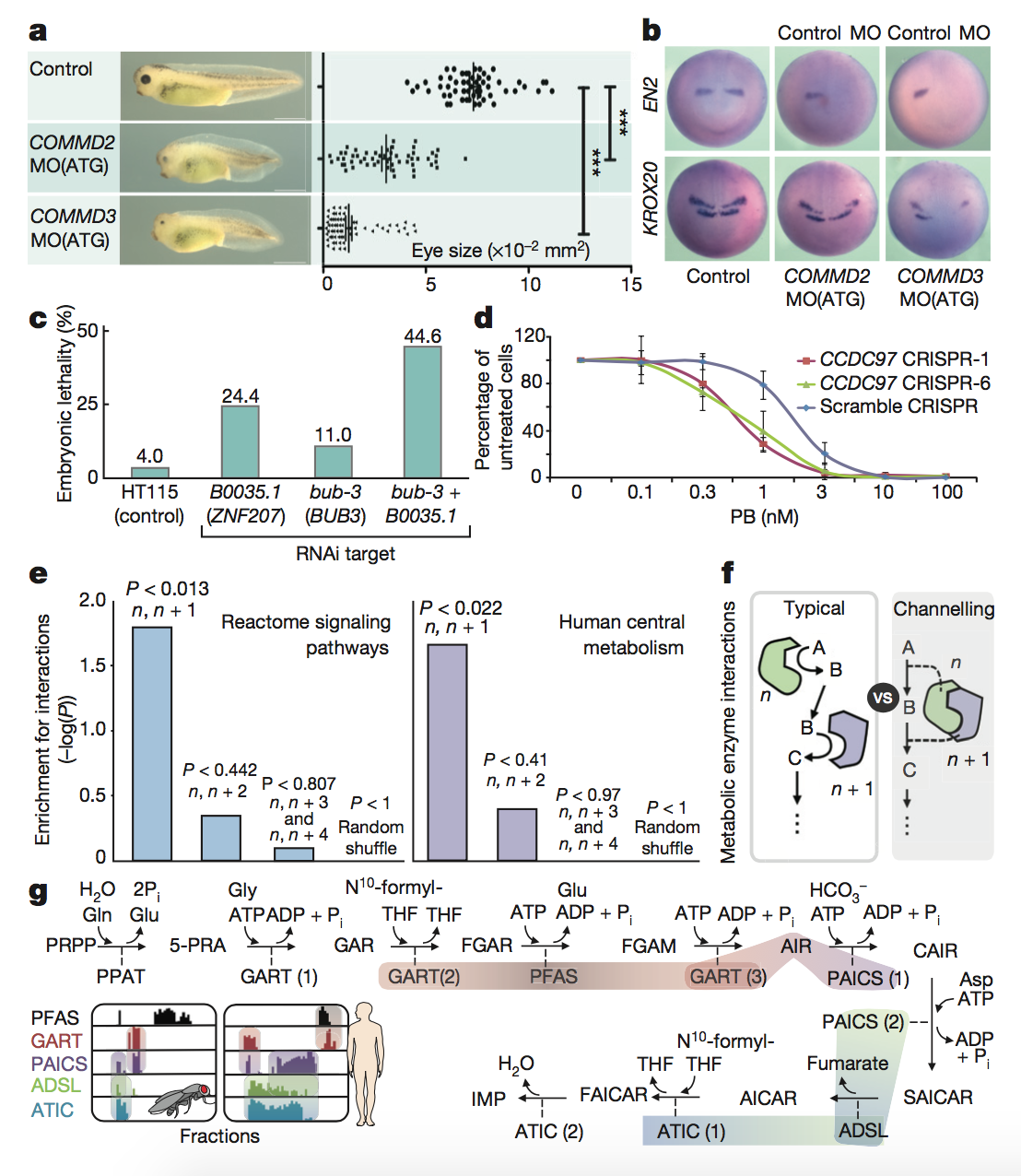
Figure 5: In
this final figure, the authors functionally
validated a number of the interactions that they had identified
previously in
the paper. They first focused on a novel complex, Commander, a large
multi-subunit protein in which the function of many subunits was
previously
unknown. They found that knocking down expression of the COMMD2 and
COMMD3
subunits of this protein led to impaired head and eye development (panel
A) as
well as impaired brain development and neural patterning (panel B) in
tadpoles.
Next, the authors focused on the metazoan-specific PPI between BUB3 and
ZNF207,
which are involved in the proper attachment of chromosomes to spindles
during
cell division. They found that RNAi knockdown of either the C.
elegans ZNF207 ortholog alone or BUB3
and ZNF207 together led
to
increased lethality for developing embryos. This had previously been
seen in
humans but not in worms (panel C). The authors then looked at mixed
protein
complexes. They found that knocking down the spliceosomal component
involved in
recognition of the branch site of unprocessed mRNA led to slower growth
and
increased sensitivity to a splicing inhibitor (panel D).
Finally, the authors
took a
network approach to these functional analyses. They found that their
fractionation data were significantly enriched for proteins that
interacted at
sequential steps in signaling pathways (panel E), which makes sense
because
these proteins would have to interact during signaling. Consecutive
interactions were also seen for widely conserved biosynthetic pathways
such as
purine synthesis (panel G), which involves metabolic channeling (when
one
enzyme passes its metabolic product directly to the active site of the
next
enzyme), as seen in panel F.
key
takeaways:
The authors’ method can be
used to identify functionally relevant PPIs as well as important network
interactions. The conservation of these PPIs can also be analyzed.
Reference:
Wan C, et al. 2015. Panorama of ancient metazoan macromolecular
complexes. Nature. 525:339-344. Available from Macmillan
Publishers.
My
Page
Genomics Page
Biology Page
Email
Questions or Comments: phparrish@davidson.edu
© Copyright 2016 Department of Biology, Davidson College, Davidson, NC
28035