How do you sequence a whole genome?
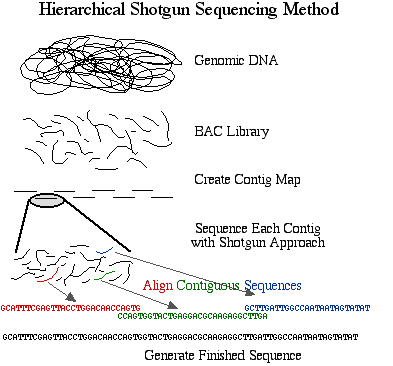
There are two general strategies for sequencing a complete genome. The method preferred by the Human Genome Project is the hierarchical shotgun sequencing method. In this approach, genomic DNA is cut into pieces of about 150 Mb and inserted into BAC vectors, transformed into E. coli where they are replicated and stored. The BAC inserts are isolated and mapped to determine the order of each cloned 150 Mb fragment. This is referred to as the Golden Tiling Path. Each BAC fragment in the Golden Path is fragmented randomly into smaller pieces and each piece is cloned into a plasmid and sequenced on both strands. These sequences are aligned so that identical sequences are overlapping. These contiguous pieces are then assembled into finished sequence once each strand has been sequenced about 4 times to produce 8X coverage of high quality data.
Figure 1. Schematic diagram of sequencing strategy used by the publicly funded Human Genome Project. The DNA was cut into 150 Mb fragments and arranged into overlapping contiguous fragments. These contigs were cut into smaller pieces and sequenced completely.
The method developed and preferred by Celera is simply called shotgun sequencing. This approach was developed and perfected on prokaryotic genomes which are smaller in size and contain less repetitive DNA. Shotgun sequencing randomly shears genomic DNA into small pieces which are cloned into plasmids and sequenced on both strands, thus eliminating the BAC step from the HGP's approach. Once the sequences are obtained, they are aligned and assembled into finished sequence.
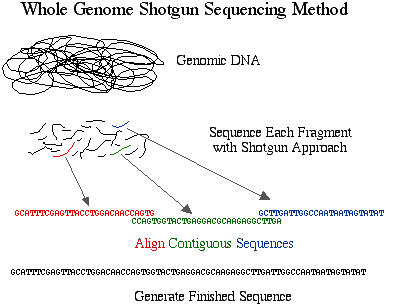
Figure 2. Schematic diagram of sequencing strategy used by Celera. The DNA was cut into small pieces and sequenced completely. These fragments were organized into contigs based on overlapping sequences.
The advantage to the hierarchical approach is sequencers are less likely to make mistakes when assembling the shotgun fragments into contigs as long as full chromosomes. The reason is that the chromosomal location for each BAC is known, and there are fewer random pieces to assemble. The disadvantage to this method is time and expense. The shotgun method is faster and less expensive, but it is more prone to errors due to incorrect assembly of finished sequence. For example, if a 500 kb portion of a chromosome is duplicated and each duplication is cut into 2kb fragments, then it would be difficult to determine where a particular 2 kb piece should be located in the finished sequence since it occurs twice. You might think, "who cares since they're duplicates?" But duplications seldom retain their original sequences; they tend to drift over time. So a small region may be retained while other parts may mutate. This might create overlapping sequences for small pieces that are located several hundred kb apart on the chromosome.
Which method is better? It depends on the size and complexity of the genome. With the human genome, each group believes its approach to be superior to the other. We only have draft sequences and each has gaps and unfinished regions so it is not possible to say for sure. It is worth mentioning that Celera had access to the HGP data but the HGP did not have access to the Celera data. Furthermore, since the Celera data is not freely available, most investigators will use the HGP sequence for further research. Therefore, we may never know which method "won".
![]()
![]()
© Copyright 2002 Department of Biology, Davidson College, Davidson,
NC 28036
Send comments, questions, and suggestions to: macampbell@davidson.edu