This page was produced as an assigment for an
undergraduate course at Davidson College.
Chronic Granulomatous Disease
Normal oxygen-dependent antimicrobial
molecule production
The NADPH oxidase complex in phagocytic cells is a multiprotein complex
with both membrane-bound and cytosolic components. Cytochrome b558
is the membrane-bound component of the NADPH oxidase complex. It
is a heterodimer, 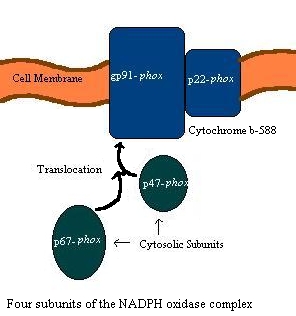 comprised
of a 22 kDa a subunit (named p22-phox)
and a 91 kDa b subunit (named gp91-phox).
The 570 amino acid gp91-phox is extensively glycosylated, while
the 195 amino acids of p22-phox are not. It is thought that
one heme group binds to the cytochrome b heterodimer, and an FAD binding
site is thought to be located in the gp91-phox subunit. The
two cytosolic components of the NADPH oxidase complex are p47-phox
and p67-phox. Less is known about these components, but they
are thought to be regulatory and not enzymatically active, and they have
SH3 domains that may interact with the cytoskeleton to put oxidase assembly
in the proper place. A small G-protein may also be associated with
the NADPH oxidase complex (Forehand
et al., 1995).
comprised
of a 22 kDa a subunit (named p22-phox)
and a 91 kDa b subunit (named gp91-phox).
The 570 amino acid gp91-phox is extensively glycosylated, while
the 195 amino acids of p22-phox are not. It is thought that
one heme group binds to the cytochrome b heterodimer, and an FAD binding
site is thought to be located in the gp91-phox subunit. The
two cytosolic components of the NADPH oxidase complex are p47-phox
and p67-phox. Less is known about these components, but they
are thought to be regulatory and not enzymatically active, and they have
SH3 domains that may interact with the cytoskeleton to put oxidase assembly
in the proper place. A small G-protein may also be associated with
the NADPH oxidase complex (Forehand
et al., 1995).
In the resting state of the cell, these components are disassociated.
However, when a phagocytic cell is activated by opsonized pathogen or other
stimuli, the subunits translocate and form a complex that eventually produces
the superoxide radical (O2-) (Forehand et al.,
1995). Upon stimulation, receptors of the signal initiate the intracellular
cleavage of PIP2. This begins a cascade that causes an
increase in intracellular Ca2+, which is thought to promote
the association of the cytosolic components of the NADPH Oxidase complex
with cytochrome b588 (OMIM, 1999). With the complex assembled,
the cell is prepared to undergo the respiratory burst. In this process,
NADPH oxidase first uses oxygen to produce superoxide, O2-.
Superoxide dismutase then catalyzes the reacation between H+
and superoxide to produce H2O2, which can also react
with O2- to produce the hydroxyl radical .OH.
H2O2 can also react with halide ions to produce hypochlorous
acids, a reaction catalyzed by myeloperoxidase. Hypochlorite can
interact with ammonia or amines to produce chloramines. It is thought
that other antimicrobials are formed in this process, as well. Thus,
the reactions of the respiratory burst serve to produce precursors to potent
microbicides (Forehand et al., 1995).
Defects in Phagocyte Function
Caused By CGD
Chronic granulomatous disease is the name given to a collection of inherited
genetic mutations that disrupt the NADPH oxidase complex. There are
four basic types of CGD, grouped by which of the four subunits (gp91-phox,
p22-phox, p47-phox, p67-phox) is affected by the mutation.
The end result of all mutations is a misfunctioning oxidase system that
produces little or none of the normal microbicidal molecules.
Approximately 60% of CGD patients have the X-linked form of CGD.
This form involves a mutation in the CYBB gene encoding gp91-phox. The
type of mutation involved varies, and includes deletions, frameshifts,
and substitution of stop codons or other active codons for amino acid codons.
Within this class of CGD, there are three phenotypes. Individuals
exhibiting the X91o phenotype have no cytochrome b588
and show no oxidase activity. Large deletions, frameshift mutations,
and nonsense mutations (substitution of a stop codon for a coding codon)
in the CYBB gene were most often observed to cause this phenotype.
Missense mutations (substitution of codons for one amino acid for those
of another) were the main cause of the X91- phenotype, characterized
by low cytochrome b588 and oxidase function levels. Some
rare cases of the X91- phenotype will have no oxidase activity
in about 90% of their neutrophils, but normal activity in the others.
Finally, patients with the X91+ phenotype, which is also most
often a result of missense mutations, will have normal levels of cytochrome
b588 but no oxidase activity (Rae et al., 1998).
The remaining 40% of patients have an autosomal form of the disease.
Almost all are autosomal recessive (Gallin and Malech, 1990). The
most frequent autosomal recessive mutations are in the genes encoding p47-phox
on
chromosome 7, NCF1. Unlike the X91 forms of the disease, mutations in p47-phox
are not diverse; they can be the result of a deletion in exon 2 that gives
a stop codon down the line, or a subsitution of guanine for alanine in
nucleotide 179 that causes the substitution of alanine for threonine in
the protein. Mutations in CYBA, the gene encoding p22-phox,are
more diverse, but all result in normal levels of cytochrome b588
without oxidase function. The mutations for p67-phox, encoded
in gene NCF-2 on chromosome 1, are not as well understood as the others
(Forehand et al., 1995).
Clinical Presentation and Diagnosis
of CGD
As a result of the reduced microbicidal activity of phagocytes caused
by the mutations of CGD, patients who have the disease
 have an increased
suceptibility to infection. Most individuals will experience symptoms
by age 3, and 95% of all cases are diagnosed by age 5. Approximately
half of those patients will live to 20 (Liese et al., 1996).
Patients with CGD have frequent infections with Staphylococcus aureus
and Aspergillus fumigatus and recurring episodes of pneumonia, abscesses
in the lung, liver, and perianal area, gastrointestinal problems, and skin
infections (Gallin and Malech, 1990). Areas normally well-protected
by phagocytes, including the spleen, liver, gastrointestinal tract, and
urogenital tract, are particularly suceptible to infection (Forehand et
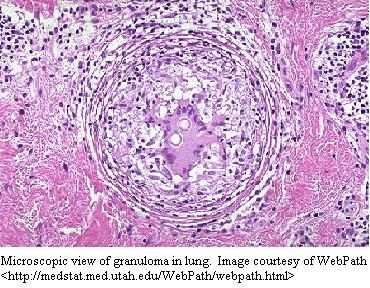
al., 1995). The characteristic chronic granulomas form because
of chronic infections and inflammation. They consist of a central
area of macrophages, some fused together to form multinucleated giant cells,
surrounded by T-cells and some plasma cells (Janeway et al., 1999).
Granulomas can further exacerbate problems by causing constrictions or
obstructions in the gastrointestinal and urogenital tracts (Gallin and
Malech, 1990). Infections easily become serious or life-threatening,
and one study showed patients with CGD spent an average of 41 days per
year in the hospital (Study Group, 1991). While CGD does not directly
cause death, premature death is often the result of an uncontrollable infection.
have an increased
suceptibility to infection. Most individuals will experience symptoms
by age 3, and 95% of all cases are diagnosed by age 5. Approximately
half of those patients will live to 20 (Liese et al., 1996).
Patients with CGD have frequent infections with Staphylococcus aureus
and Aspergillus fumigatus and recurring episodes of pneumonia, abscesses
in the lung, liver, and perianal area, gastrointestinal problems, and skin
infections (Gallin and Malech, 1990). Areas normally well-protected
by phagocytes, including the spleen, liver, gastrointestinal tract, and
urogenital tract, are particularly suceptible to infection (Forehand et
al., 1995). The characteristic chronic granulomas form because
of chronic infections and inflammation. They consist of a central
area of macrophages, some fused together to form multinucleated giant cells,
surrounded by T-cells and some plasma cells (Janeway et al., 1999).
Granulomas can further exacerbate problems by causing constrictions or
obstructions in the gastrointestinal and urogenital tracts (Gallin and
Malech, 1990). Infections easily become serious or life-threatening,
and one study showed patients with CGD spent an average of 41 days per
year in the hospital (Study Group, 1991). While CGD does not directly
cause death, premature death is often the result of an uncontrollable infection.
Diagnosis of CGD involves several in vitro phagocyte-function
assays. The standard screening test for CGD is a nitroblue tetrazolium
(NBT) test, in which neutrophils are incubated with yellow NBT and examined
microscopically for color changes. When NBT is reduced by O2-
it
changes color to blue and becomes less soluble. Normal neutrophils
will readily reduce NBT; CGD patients will show little or no color changes
in their cells. Other assays can measure superoxide production, oxygen
consumption, chemiluminescence (an effect caused by the respiratory burst),
and hydrogen peroxide production. If these tests indicate abnormalities,
genetic testing can be done for a definite diagnosis and to determine which
form of CGD the patient has (Forehand et al., 1995).
Current and Future Treatments
for Individuals with CGD
Treatment of CGD has centered around treatment of the infections that
arise in CGD patients. Many CGD individuals are on prophylactic antibiotics,
generally a combination of sulfamethoxazole and trimethoprim (TMP-SMX).
These drugs help eliminate S. aureus infections, prolonging the
time between serious infections (Gallin and Malech, 1990). The cytokine
interferon gamma (IFN-g) can also promote killing
of S. aureus and Aspergillus fumigatus. Early studies
with IFN-g seemed to indicate an enhanced respiratory
burst, but later studies did not find evidence of this (Forehand et
al., 1995). Although the exact mechanism by which IFN-g
improves
pathogen killing is unknown, the benefit of treatment is undeniable.
In an extensive study of 165 patients with CGD, IFN-g
treatment
resulted in a 67% reduction in the risk of serious infection (Study Group,
1991). Granulocyte transfusions can help in a persistent infection,
but toleration of the transfusion is an issue (Forehand et al.,
1995).
Direct cures for CGD would involve changing the precursors for phagocytic
cells to cells with proper function. Bone marrow transplants have
been used with mixed results. The obvious risk is transplant rejection
and the risks associated with further immunocompromising patients (Forehand
et
al., 1995). Some attempts have been made to introduce the missing
oxidase components into the cells using viruses. These techniques
encode the missing protein in the viral genome, introduce the virus to
the cells, and allow normal retrovirus mechanisms to produce the protein
(OMIM, 1999). A new possibility arises with stem-cell research.
A patient's own stem cells could be removed and genetically altered before
replacing, or a stem cell transplant from a related individual could be
used as a cure. These procedures have the benefit of requiring less
immunosuppression (Weaver, 2000).
References and Further Reading
Dinauer MC, Orkin SH, Brown R, Jesaitis AJ, Parkos CA. The glycoprotein
encoded by the X-linked chronic granulomatous disease locus is a component
of the neutrophil cytochrome b complex. Nature 1987 25 June;327:717-20.
Forehand JR, Nauseef WM, Curnutte JT, Johnston RB. 1995.
Inherited Disorders of Phagocyte Killing. In: Scriver CR, Beaudet
AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of
Inherited Disease. 7th ed. New York: McGraw-Hill, Inc.
Health Professions Division. p 3995-4026.
Gallin JI, Malech HL. Update on Chronic Granulomatous Diseases
of Childhood: Immunotherapy and Potential for Gene Therapy.
JAMA 1990 16 March;263:1533-7.
[Study Group] The International Chronic Granulomatous Disease Cooperative
Study Group. A Controlled Trial of Interferon Gamma to Prevent Infection
in Chronic Granulomatous Disease. New Engl J Med 1991 21 February;324:509-16.
Janeway CA, Travers P, Walport M, Capra JD. Immunobiology:
The Immune System in Health and Disease. 4th ed. New York:
Garland Publishing; 1999. p 301-3.
Liese JG, JendrossekJansson A, Petropoulou T, Kloos S, Gahr M, Belohradsky
BH. Chronic Granulomatous Disease in Adults. Lancet 1996 27
January;347:220-3.
[OMIM] Online Mendelian Inheritance in Man. 1999 Mar 24.
306400 Granulomatous Disease, Chronic; CGD. <http://www3.ncbi.nlm.nih.gov:80/htbin-post/Omim/dispmim?306400>.
Accessed 2000 Apr 17.
Rae J, Newburger PE, Dinauer MC, Noack D, Hopkins PJ, Kuruto R, Curnutte
JT. X-Linked Chronic Granulomatous Disease: Mutations in the
CYBB
Gene Encoding the gp91-phox Component of Respiratory-Burst Oxidase.
Am J Hum Genet 1998 62:1320-31.
Teahan C, Rowe P, Parker P, Totty N, Segal AW. The X-linked chronic
granulomatous disease gene codes for the b-chain
of cytochrome b-245. Nature 1987 25 June;327:720-1.
Weaver J. 2000 27 March. Married!!!!!Oh MiGosh!!!!! [Personal
Email]. Accessed 2000 28 March.
Comments? Email me.
Go back to front page.
Link to BIO 307 Immunology
home page
Link to Davidson College home
page.