![]()
Rheumatoid Arthritis

The hand of a rheumatoid arthritis patient is pictured above (courtesy of HeliosHealth.com)
(Click on photo to link to HeliosHealth.com)
![]()
![]() Introduction to Autoimmune Disease and Rheumatoid Arthritis
Introduction to Autoimmune Disease and Rheumatoid Arthritis
Autoimmunity
is caused by an immune response to self antigens, which normally should
not ellicit an immune resoponse (Janeway et al., 1999). The
purpose of a normal immune response is to completely eliminate invading
antigen from the body, and the response stops once the foreign antigen(s)
have been completely cleared. However, since the self antigen that
elicits an autoimmune response is an intrinsic component of the body and
usually impossible to eliminate, and the immune response is sustained.
Such sustained immune responses lead to chronic inflammation and injury
to tissues, which can become lethal (Janeway et al., 1999).
Rheumatoid arthritis
(RA), characterized by painful swelling, deformity, and deterioration of
the joints, affects 0.5 - 1% of the worldwide population with an age of
onset between 40 and 50 years old. RA patients generaly have a life
expectancy that is 3-18 years shorter than wild type individuals (Strand,
1999), and women develop the disease three times as of often as men (Janway
et
al., 1999). The disease is charaterized as a type IV T-cell-mediated
disease in which specifically activated T-cells that respond to self peptide:self
MHC complexes are the main contributors to pathogenesis. The autoimmune
response is elicited by an unknown synovial
(i.e. in the fluid surrounding the joints) joint antigen is the autoantigen
(Janeway et al., 1999). Since there is no cure for RA,
the goal of treatment and therapy is to control the underlying inflammatory
process and maintain or improve joint function (Strand, 1999).
![]() Pathogenesis of Rheumatoid Arthritis
Pathogenesis of Rheumatoid Arthritis
The autoimmune
response in RA is caused by the specific activation of self-reactive Th1
cells, which infiltrate the synovial fluid surrounding the joints (Wagner
et
al., 1998). Activated T-cells release lymphokines that initiate
the inflammatory response by recruiting lekocytes and macrphages to the
joint area, causing cartilage damage and destruction of the joint (Janeway
et al., 1999). Rheumatoid factor,
is an IgM anti-IgG autoantibody, participates in a T-cell dependent B-cell
reponse against the Fc portion of IgG. The resulting IgM:IgG immune
complexes cause some tissue damage (Janeway et al., 1999).
Pro-inflammatory cytokines play a key role in RA pathogenesis, two of the
most important being tumor necrosis factor-a
(TNF-a) and interleukin-1b
(IL-1b). Both cytokines are secreted by
synovial macrophages and stimulate the proliferation of synovial cells,
the production of collagens, and the expression of adhesion molecules on
the endothelium, which enhances the recruitment of more lymphocytes (Strand,
1999). There are at least six cytokines (including TNF-a)
present in the the synovial fluid of RA patients that are known to regulate
interleukin-1
(IL-1), which has been observed to induce the destruction of bone and cartilage
(Feldmann, 1999). Current therapies that target cytokines include
soluble-receptor antagonists and cytokine antibodies (Strand, 1999).
Abnormalities in the T-cell repertoire of RA patients include the presence
of clonal T-cell populations. The
formation of clonal T-cell populations causes "repertoire
contraction," which is a reduction in the
diversity of the T-cell receptor (TCR) b-chain
sequences observed in circulating CD4 T-cells (Wagner et al., 1998).
T-cell clones are produced as a result of antigen-specific stimulation
of T-cells. Some abnormal features of the clones are that they expand
in size, are autoreactive to ubiquitous proteins, do not express CD28,
and do not require co-stimulation in order to secrete lymphokines (Wagener
et
al., 1998). The production of T-cell clone populations causes
the observed TCR-b chain repertoire reduction
because the clones are present at a higher frequency than other TCR b-chains.
Under nonstimulated conditions, as in wild type individuals, the CD4 T-cell
repertoire is very diverse, with each TCR-b
chain present at a low frequency. In addition, some tissue-infiltrating
lymphocytes are organized into clusters or follicles that resemble germinal
centers. The similarity of these follicles to lymphoid tissue supports
the concept that rheumatoid synovitis
is the consequence of an antigen-driven response.
Another abnormailty found in the CD4+ T-cells of RA patients is that the
expression of CD40 ligand (CD40L) is significantly higher than in wild
type individuals (Berner et al., 2000). CD40, the glycoprotein
receptor for CD40, belongs to the tumor necrosis factor receptor (TNFR)
family and is expressed on a wide variety of cells such as B-cells, vascular
endothelial cels, monocytes and macrophages, and dendritic cells.
Binding of the CD40 on CD4+ T-cells to CD40L produces signalling that is
crucial in cell-mediated as well as humoral immune responses. T-cells
expressing CD40L that infiltrate the synovial fluid interact with fibroblasts
expressing CD40, which induces fibroblast proliferation, increased recruitment
of inflammatory cells, and production of TNF-a.
In addition, the production of interleukin-2
(IL-2) by dendritic cells, which is required for the initiation of Th1
cell responses, is regulated by the CD40-CD40L interaction. Since
RA is a Th1 cell-mediated disease, the CD40-CD40L interaction
may be an important pathogenic pathway (Berner et al., 2000).
The increased expression of CD40L is still observed 5-12 years after disease
onset, indicating augmented and prolonged T-cell activation.
![]() Treatments
Treatments
There has
been a recent boom in the development of cytokine therapies for RA, which
include be soluble-receptor antagonists or cytokine antibodies. It
was originally thought that cytokines would be a poor target for therapy
since it was assumed that other cytokines would be enough to maintain the
immune response if one cytokine was blocked. However, research has
shown that blocking TNF-a inhibited IL-2 activity,
despite the presence of other IL-2-regulating cytokines in the synovial
fluid (Feldmann et al., 1999). This led to the hypothesis
that pro-inflammatory cytokines in diseased tissue are co-regulated, not
regulated independently, with the key coordinators in inflammation being
TNF-a and IL-2. The resulting overlap
in the propertied of pro-inflammatory cytokines is termed "redundancy"
(Feldmann et al., 1999), and the discovery of redundancy has led
to increased interest in cytokine therapies.
Two types
of drugs used in the treatment of RA are non-steroidal
anti-inflammatory drugs (NSAIDs) and disease-modifying
anti-rheumatic drugs (DMARDs). Leflunomide, etanercept (Enbrel),
infliximab, and protein A immunoadsorption column (used with plasmapheresis
therapy) are four recently-developed RA tratments. Leflunomide
is a DMARD therapy that inhibits de novo synthesis of pyrimidine,
which causes reversible cell-cycle arrest in rapidly dividing cells such
as activated lymphocytes (Strand, 1999). Thus, leflunomide is an
anti-proliferative agent. Etanercept
(a.k.a. Enbrel) was the first biologic agent
to be approved for RA treatment. It is a recombinantly-produced form
of soluble type II TNF-a receptor (given in
25-mg injections twice weekly), which binds to free-floating TNF-a,
thereby preventing it from binding to a cell-surface receptor and initiating
a cytokine cascade (Strand, 1999). The animation below (Figure 1)
illustrates the mechanism of Enbrel.
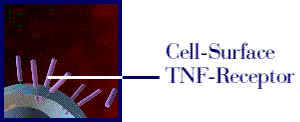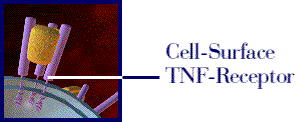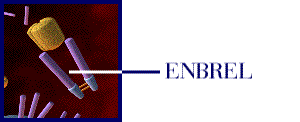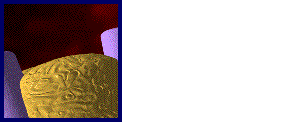
Figure 1: The above movie, courtesy of Immunex, Inc., illustrates how Enbrel inhibits the binding of TNF to the cell-surface TNF receptor, thereby preventing the inflammation response associated with rheumatoid arthritis. Permission pending from www.immunex.com. (link to image source)
Infliximab, a chimeric IgG1 monoclonal TNF-a antibody, is also administered intravenously and has been shown to provide rapid, long-lasting benefits. The protein A immunoadsorption column, for use with plasmapheresis therapy, is the first device approved for the treatment of RA. Protein A, known to bind to IgG, is a staphylococcus bacteria-derived protein that is covalently bound to a silica matrix in a column. It is thought that plasmepheresis using this type of column allows for binding and in vivo clearance of immune complexes (Strand, 1999).
![]() References
References
Berner B, Wolf G, Hummel K, Müller G, Reuss-Borst MA. 2000. Increased expression of CD40 ligand (CD154) on CD4+ T cell as a marker of disease activity in rheumatoid arthritis. Annals of Rheumatic Disease 59: 190-195.
Feldmann M, Bondeson J, Brennan FM, Foxwell BMJ, Maini RN. 1999. The rational for the current boom in anti-TNFa treatment. Is there an effective means to define theraputic targets for drugs that provide all the benefits of anti-TNFa and minimise hazards? Annals of Rheumatic Disease 58: 127-131.
Helios Health, Inc. 2000 April 12. Arthritis. <http://www.helioshealth.com/arthritis/rheumatoid.html> Accessed 2000 April 20.
Immunex, Inc. Breakthrough Science. <http://www.enbrelinfo.com/patient/html/breaksci.html> Accessed 2000 April 17.
Janeway C, Travers P, Walport M, Capra JD. 1999.
Immunobiology: The Immune System in
Health and Disease. New York, NY: Elsevier
Science Ltd./Garland Publishing.
Strand, Vibeke. 1999. Recent Advances in the Treatment of Rheumatoid Arthritis. Clinical Cornerstone2: 38-47.
Wagner UG, Koetz K, Weyand CM, Goronzy JJ. 1998.
Perturbation of the T-cell repertoire in rheumatoid arthritis.
Proc.
Natl Acad. Sci. 95: 14447-14452.
Back to Top
Back to Lymphotoxin homepage