This web page was produced as an assignment for
an undergraduate course at Davidson College.
Adenosine deaminase deficiency and treatment
by Christopher D. Lee
Return to my Immunology Homepage
Tapasin
Adenosine deaminase
ADA deficiency
Treatment
Sources
Figure 1: A Chime view of adenosine deaminase.
This is ADA1, a 349 amino-acid enzyme. This structure has a His238
Ala Mutation. Authors: D.K.Wilson & F.A.Quiocho. From MedLine,
PubMed. Source.
Can't see the figure? Download
Chime.
The enzyme adenosine deaminase
ADA deficiency
Treatment
Sources
Adenosine deaminase (Fig. 1), or ADA (also referred
to as adenosine aminohydrolase, EC 3.5.4.4), is a ubiquitous enzyme which
appears to be particularly important in the development of thymocytes.
ADA converts adenosine into inosine (Fig. 2) and converts
deoxyadenosine (dAdo) into deoxyinosine, both through the hydrolysis of
the purine amino group (Benveniste and Cohen, 1995). ADA is present
in all tissues, but has much higher activity in lymphocyte development,
perhaps due to its direct association with CD26, which is exhibited on
activated T cells (Tsuboi, et al., 1995). ADA activity is
particularly high in thymocytes of the thymic cortex, but drops off rapidly
in the medulla (Resta, et al., 1997). There are two enzymes
which carry out ADA activity, called ADA1 and ADA2. ADA1 (Fig.
1), a 40 kD monomeric protein with a 200 kD, noncatalytic combining
protein, is responsible for about 90% of adenosine deamination. ADA2
is somewhat larger at 110 kD, appears to play a general adenosine deamination
role in serum (Tsuboi, et al., 1995).
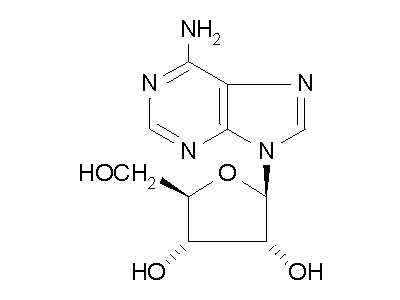

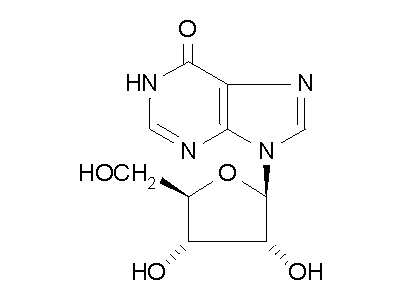
Figure 2: ADA hydrolysis of adenosine into inosine.
ADA also acts on deoxyadenosine, which has no 2' hydroxy group on the ribose
unit, and dATP, which also has three phospate units on the 5' hydroxy group.
Hydrolysis cleaves the amino group, resulting in an amide, and releasing
an ammonia molecule.
ADA Deficiency
Adenosine deaminase
Treatment
Sources
These conversions are beginnings of important degratory pathways of
purines in cells. The main biochemical consequences are:
-
Individuals that are ADA deficient have abnormal accumulations of dAdo
and dATP (deoxyadenosine triphosphate), which is the phosphorylated end-product
of of dAdo.
-
These accumulations result in the inactivation of an enzyme called S-adenosylhomocysteine
hydrolase (SAH), whose substrate accumulation inhibits certain methylations
of nucleic acids, proteins, and lipids (Benveniste and Cohen, 1995).
-
High levels of dAdo are also responsible for an inhibition of ribonucleotide
reductase, causing an inbalance of the deoxynucleotides (dNTP).
-
This imbalance, in addition to the enzyme inhibitions, leads to impairment
of DNA synthesis and repair in T lymphocytes (Benveniste and Cohen, 1995).
It appears that thymocytes are most affected by ADA deficiency during maturation
in the cortex of the thymus, specifically at the transitional CD8low
and CD4+CD8+ stages, which coincide with recombination
of the T cell receptor (TCR) (Benveniste and Cohen, 1995). At this
stage, expression of bcl-2 is low, so the thymocytes are particularly
succeptible to apoptosis. The impairment of DNA repair due to the
imbalanced levels of dNTPs leads to an accumulation of DNA breaks in the
cell genome, which eventually triggers apoptosis in the bcl-2 low
environment, also dependent on expression of a protein called p53 (Benveniste
and Cohen, 1995). dATP toxicity also leads to exhaustion of ATP and
NAD (Gangi-Peterson, et al., 1999), further inhibiting cell function.
Improperly functioning ADA, subsequent dAdo, dATP, and dNTP imbalances,
as well as the impairment of S-adenosylhomocysteine hydrolase contribute
to apoptosis of developing thymocytes (Gangi-Peterson, et al., 1999),
resulting in loss of T cell function and leading to severe combined immunodeficiency
(SCID).
Symptoms for ADA deficiency-induced SCID are usually immediately seen
in affected infants, since the disorder is genetic, but there have been
cases of mild ADA deficiency which were not been detected until older childhood
and even adulthood (Ozsahin, et al., 1997). Complete ADA deficiency
results in fatal infantile onset syndrome of SCID. Even in milder
cases, T-cell function is severely depressed, and antibody responses are
barely produced, resulting in a highly immunocompromised individual (Ochs,
et
al., 1992). Patients with the disorder exhibit growth retardation,
are susceptible to opportunistic infections, lymphopenia, and defective
cellular and humoral immune responses (Ozsahin, et al., 1997).
At least 40 alleles have been identified to cause ADA deficiency.
There appear to be specific locations on the gene which are unusually succeptible
to mutations in the characterized alleles, so called hot-spot mutations
(Hirschhorn, et al., 1990).
It has been noted above that cortical thymocytes exhibit high levels
of ADA activity at the CD8low and CD4+CD8+
stages of development, which coincide with recombination of the T cell
receptor (TCR). One of the consequences of dAdo and dNTP imbalances
at this stage is that V(D)J recombination is affected by the imbalance
of nucleotides available for the non-template encoding regions, or N-regions,
during assembly of the V(D)J joints of TCR chains and immunoglobulins.
N-regions are typically G-C rich, but cells treated with dAdo, which simulates
ADA deficiency, exhibit N-regions with significant A-T nucelosides (Gangi-Peterson,
et
al., 1999). This tendency was also found in isolated cells of
ADA deficient patients, and the recombination frequency of Igs and TCRs
are significantly reduced (Gangi-Peterson, et al., 1999).
The specific effects of this change are unknown, but there is potential
for disruption of the V(D)J recombination event or for a decrease in the
diversity of immunoglobulins and T cell receptors, which might constrain
immune responses. TdT is the enzyme that adds the N-nucleotides in
recombination, and TdT knockout mice interestingly have normal B cell and
T cell responses (Gangi-Peterson, et al., 1999), indicating that
the A-T rich N-regions may have a more deleterious effect on development
than no N-regions at all.
Treatment Adenosine
deaminase
ADA deficiency
Sources
Treatment of ADA deficiency is possible through 3 main routes:
-
Bone marrow or stem cell transplants from a haploidentical donor is available
for a minority of patients (Bordignon et al., 1995). Since
the disorder is genetic, haploidentical donors who are not ADA deficient
themselves are harder to find than for leukemia or another bone marrow
transplant scenario. Bone marrow transplants can be performed to
correct general cases of SCID, and the new marrow cells contain the appropriate
genes, including ADA+, to develop normal T and B cells, reconstituting
immune function (Bordignon, et al., 1995; Haddad, et al.
1999).
-
Enzyme therapy can directly add missing ADA. This can occur through
a transfusion of irradiated red blood cells. Patients on this regime
experienced equally depressed antibody responses as untreated ADA deficient
patients (Ochs, et al., 1992). Direct enzyme injections are
a better method of introducing ADA to the patient. There have also
been several studies which have discovered efficient methods of obtaining
high yield human ADA by transfecting various cells and insect larvae with
human ADA cDNA (Medin, et al., 1990). In treatment, human
or bovine ADA is covalently attached to polyethylene glycol (PEG), which
blocks access of degrative enzymes to ADA, lengthening the plasma half-life
from a few minutes to 24 hours. Weekly injections of PEGilated
ADA usually reverse the main consequences of genomic ADA deficiency, as
lymphocyte levels and proliferative responses to antigen normalize (Bordignon,
et
al., 1995). Levels of dAdo and dATP are also reduced, as expected
in an effective treatment. Enzyme therapy does not work for all patients.
In some individuals, T cell function increases to mitogens in vitro, but
the patient is not able to consistently mount immune responses to select
antigens (Blaese, et al., 1995), resulting in PEG-ADA treatment
failure.
-
Somatic gene therapy can create functional ADA+ T cells.
ADA deficiency was the first disorder to be treated by gene therapy (Bordignon,
et al., 1995). The initial targets for genetic manipulation were
bone marrow (BM) stem cells and peropheral blood lymphocytes (PBL).
Vectors expressing human ADA cDNA, 1.5 kbp (Blaese, et al., 1995)
with their own promoters can be transfected into BM stem cells and PBLs
in vitro. 6 months after gene therapy ended, vector-derived DNA is
found in PBLs, and levels of leukocytes and bone marrow progenitors increases.
ADA activity is substantially improved, and short term (6-12 months) immunity
is effectively restored (Bordignon, et al., 1995). These experiments
also confirm that genetically modified cells with properly functioning
ADA have a selective advantage over endogenous ADA deficient cells during
development. Even growth was normalized in some patients whose growth was
not improved with PEG-ADA treatment (Bordignon, et al., 1995).
Since T cells suffer most from ADA deficiency, and T cells were
essentially the only cells whose ADA activity rose under gene therapy,
the possibility of T-cell directed gene modification was also investigated
(Blaese, et al., 1995). T cells are obtained through apheresis,
and they proliferate in culture, while they are transfected with human
ADA cDNA from a retrovirus. After 9-12 days, the T cells are reintroduced
into the patient (Blaese, et al., 1995). The ability to transfect
cells in vitro offers an advantage that the patient is not injected with
a virus, which could have uncontrollable consequences. The direct
targeting also increases the effectiveness of even low gene transfer efficiency.
With T cells, even 1% gene transfer efficiency results in 109
to 1010 new T cells, which contribute to immune diversity (Blaese,
et
al., 1995).
The use of gene therapy has more ethical concerns than the other methods,
but there are key advantages to its use in this disorder. First,
the patient can continue PEG-ADA treatment, so that no treatment is withheld.
Second, live virus is not introduced into the patient, as was the case
in the recent death of 18-year old Jesse Gelsinger, who was undergoing
gene therapy for ornithine transcarbamylase deficiency (OTC), a liver disorder.
(CNN.com, December 8, 1999) Instead, T cells are isolated, transfected,
then reintroduced to the patient, avoiding any potential complications
with a virus. Third, T cells are directly targeted instead of stem
cells, and they are long-lived and proliferate well after genetic manipulation.
(Pollok, et al., 1998) This makes gene therapy an attractive
solution to the problems created by ADA deficiency, which can be used when
enzyme therapy fails.
Sources Adenosine
deaminase
ADA deficiency
Treatment
Benveniste P, Cohen A. 1995 August 29. p53 expression is required for
thymocyte apoptosis induced by adenosine deaminase deficiency. Proceedings
of the National Academy of Science 92: 8373-8377. Abstract
PDF Text
Bordignon C, Notarangelo LD, Nobili N, Ferrari G, Casatori G, Panina
P, Mazzolari E, Maggiono D, Rossi C, Servida P, Ugazio AG, Mavilio F. 1995
October 20. Gene therapy in peripheral blood lymphocytes and bone marrow
for ADA- immunodeficient patients. Science 270: 470-475.
Abstract
Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, Shearer
G, Chang L, Chiang Y, Tolstoshev P, Greenblatt JJ, Rosenberg SA, Klein
H, Berger M, Mullen CA, Ramsey WJ, Muul L, Morgan RA, Anderson WF. 1995
October 20. T lymphocyte-directed gene therapy for ADA- SCID:
Initial trial results after 4 years. Science 270: 475-480.
Abstract
Gangi-Peterson L, Sorscher DH, Reynolds JW, Kepler TB, Mitchell BS.
1999 March 15. Nucleotide pool imbalance and adenosine deaminase deficiency
induce alterations of N-region insertions during V(D)J recombination. The
Journal of Clinical Investigations 103: 833-841. Abstract
Full Text
Haddad E, Le Diest F, Aucouturier P, Cavazzana-Calvo M, Blanche S, de
Saint Basile G, Fischer A. 1999 October 15. Long-term chimerism and B-cell
function after bone marrow transplantation in patients with severe combined
immunodeficiency with B cells: a single-center study of 22 patients. Blood.
94:
2923-2930. Abstract
Full
Text
Hirschhorn R, Tzall S, Ellenbogen A. 1990 August 15. Hot spot mutations
in adenosine deaminase deficiency. Proceedings of the National Academy
of Science 87: 6171-6175. Abstract
PDF Text
Lynch CM, Clowes MM, Osborne WRA, Clowes AW, Miller AD. 1992 February
1. Long-term expression of human adenosine deaminase in vascular smooth
muscle cells of rats: a model for gene therapy. Proceedings of the National
Academy of Science 89: 1138-1142. Abstract
PDF Text
Medin JA, Hunt L, Gathy K, Evans RK, Coleman MS. 1990 April. Efficient,
low-cost protein factories: Expression of human adenosine deaminase in
baculovirus-infected insect larvae. Proceedings of the National Academy
of Science 87: 2760-2764. Abstract
PDF Text
Ochs HD, Buckley RH, Kobayashi RH, Kobayashi AL, Sorensen RU, Douglas
SD, Hamilton BL, Hershfield MS. 1992 September 1. Antibody responses to
bacteriophage phi X174 in patients with adenosine deaminase deficiency.
Blood
80: 1163-1171. Abstract
Ozsahin H, Arredondo-Vega FX, Santisteban I, Fuhrer H, Tuchschmid P,
Jochum W, Aguzzi A, Lederman HM, Fleischman A, Winkelstein JA, Seger RA,
Hershfield MS. 1997 April 15. Adenosine deaminase deficiency in adults.
Blood
89:
2849-2855. Abstract
Full
Text
Pollok KE, Hanenberg H, Noblitt TW, Schroeder WL, Kato I, Emanuel D,
Williams DA. 1998 June. High-efficiency gene transfer into normal
and adenosine deaminase-deficient T lymphocytes is mediated by transduction
on recombinant fibronectin fragments. Journal of Virology 72:
4882-4892. Abstract
Full Text
Resta R, Hooker SW, Laurent AB, Jamshedur Rahman SM, Franklin M, Knudsen
TB, Nadon NL, Thompson LF. 1997 February 15. Insights into thymic purine
metabolism and adenosine deaminase deficiency revealed by transgenic mice
overexpressing ecto-5-nucleotidase (CD73). The Journal of Clinical
Investigations 99: 676-683. Abstract
Full Text
Staff report, 1999 December 8. FDA says youth who died in gene therapy
trial should not have received the treatment. CNN.com. Available:
<http://cnn.com/1999/HEALTH/12/08/gene.therapy.hearings/>
Accessed: April 25, 2000.
Tsuboi I, Sagawa K, Shichijo S, Yokoyama MM, Ou DW, Wiederhold MD. 1995
September. Adenosine deaminase isoenzyme levels in patients with human
T-cell lymphotropic virus type 1 and human immunodeficiency virus type
1 infections. Clinical and Diagnostic Laboratory Immunology 2:
626-630. Abstract
PDF Text
My Immunology Homepage
Tapasin
More
Students' Homepages
Davidson Immunology
Davidson Biology

This page was written by Christopher
Lee. Last Updated 21 April 2000.
© Copyright 2000, Christopher Lee