This web page was produced as an assignment for an undergraduate course at Davidson College.
Upon infection with a pathogen, host leukocytes and some non-immune cells begin secreting a variety of cytokine proteins. In general, cytokines have a wide variety of effects, ranging from activation of components of the adaptive and innate immune responses and direct action on infected cells (Biron, 1998).
Interferons are a family of cytokines produced in response to viral infections. Interferons interfere with viral replication and proliferation in vitro; this interference is how they were discovered and is the source of their name (Janeway et al., 1999). Interferons (IFNs) are classified as either type I or type II. Type I IFNs include IFN-a,-b,-t, and -w, which are all monomeric; the only type II IFN is IFN-g, a dimer. Twelve different subtypes of IFN-a are produced by 14 genes, but all other IFNs are monogenic (Arduini et al., 1999). This webpage will focus on the molecular biology and immunological importance of IFN-b.
IFN-b is a globular protein consisting of 5 a helices. It has a calculated MW of 20 kDa, though it often runs on SDS-PAGE gels with an apparent MW closer to 25 kDa due to glycosylation (Arduini et al., 1999).
View a Chime image of Interferon b
This image displays IFN-b as a homodimer; this is merely an artifact of the crystallization process. In vivo, IFN-b does not dimerize. Notice also that this is an image of glycosylated IFN-b.
Chime image retrieved from National Center for Biotechnology Information (NCBI).
Double-stranded RNA (dsRNA) seems to be one of the primary intracellular signals for interferon production. Healthy cells do not normally contain dsRNA, which is often produced during viral replication and which forms the genome of many viruses (Janeway et al., 1999; Biron, 1998). The mechanism for initial activation of IFN synthesis subsequent to viral infection is currently unknown, but several transcription factors regulating the IFN-b gene have been described (Harada et al., 1998; Pitha et al., 1998).
IRF-1 and IRF-2
IRF-1 and -2 are transcription factors in the interferon regulatory factor (IRF) family which play a major role in regulation of the IFN-b gene. IRF-1 induces IFN-b transcription in virally infected cells, while IRF-2 binds competitively to the same DNA sequence and has a repressor function. These factors bind to IRF-E, a DNA binding element in the virus inducible region (VRE) of the IFN-b promoter (Harada et al., 1998; Pitha et al., 1998).
Alternate Induction Pathways
Studies indicate
that induction of INF-b does not always require
the action of IRF-1 and -2. Mice lacking the IRF-1 gene
are still able to produce IFN-b in response to viral
infection, though the mice do exhibit an impaired immune response.
It is thought that another member of the IRF family may be responsible
for IFN-b induction in this scenario (Pitha
et al., 1998).
Even in cells
with functional IRF-1 genes, induction of IFN-b
does not always require IRF-1 synthesis. The VRE region
of the IFN-b gene contains cis-acting
DNA sequences which recruit a multicomponent enhancer complex
to the VRE. This enhancer complex, termed an enhancesome,
then recruits general transcription factors to the IFN-b
promoter. This enhanceosome does contain an unknown IRF
protein, though this pathway differs from the usual IRF-1 induction
of IFN-b in that the latter pathway requires
de novosynthesis of IRF and the former does not (Pitha
et al., 1998).
IFN-b Signal Transduction
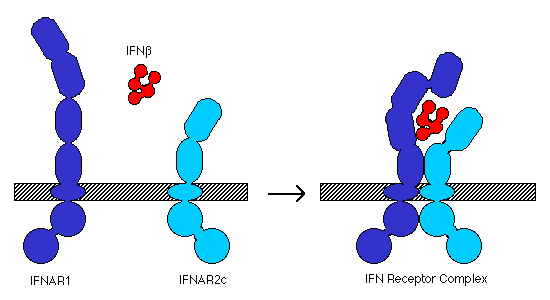
Cellular response to IFN-b is mediated by the IFN receptor, which is found on many different cell types (Biron, 1998). The receptor is a heterodimer formed by IFNAR1, a 550 residue integral membrane protein, and IFNAR2, a 487 residue integral membrane protein. The extracellular domains of IFNAR1 and 2 are composed of several fibronectin III-like repeats (IFNAR1 has 4 repeats, IFNAR2 has only 2; Arduini et al., 1999). In the presence of IFN-b, the two chains assemble into a functional receptor complex (Figure 1), which initiates the signal transduction pathway (Russell-Harde et al., 1999).
Figure 1. Model for interaction of IFN b and the type I IFN receptor components, IFNAR1 and IFNAR2c, to form a complete receptor complex. Adapted from Russell-Harde et al. (1999).
Upon assembly of the IFN receptor complex, the intracellular domains of IFNAR1 and IFNAR2 associate with two Janus-family tyrosine kinases, JAK1 and Tyk2, which transphosphorylate themselves and phosphorylate the receptors. The phosphorylated IFNAR1 and IFNAR2 then bind to STAT1 (signal transducer and activator of transcription 1) and STAT2. The STAT proteins then dimerize and migrate to the nucleus where they activate transcription of multiple genes (Arduini et al., 1999). Downregulation of the JAK/STAT pathway appears to be effected by the tyrosine phosphatase SHP-1 (SH2-containing tyrosine phosphase-1; Min et al. 1998).
Interestingly,
IFN-a and -b share the same membrane
receptor yet elicit different cellular responses. Russell-Harde
et al. (1999) showed that IFN-b binds to IFNAR1 and IFNAR2
more stably than does IFN-a. The stability
of the IFN receptor complex is thus postulated to have an effect
on the nature of subsequent signal transduction, allowing for
IFN-a and -b to elicit different responses
via the same receptor.
Immune
System Effects of IFN-b
IFN-b, acting
via STAT1 and STAT2, is known to upregulate
and downregulate a wide variety of genes, most of which are involved
in the antiviral immune response. Although most IFN responses
are induced by the presence of dsRNA, both DNA and RNA viruses
are sensitive to the effects of IFN-b (Biron, 1998).
MHC class I / TAP / Lmp2 / Lmp7
IFN-b is generally produced in response to a viral infection, and the IFN-b-dependent upregulation of these genes serves to increase presentation of viral peptides by MHC class I molecules in order to facilitate CD8 T cell recognition and destruction of infected cells. TAP (transporter associated with antigen processing) is the molecule responsible for loading peptide fragments onto MHC class I molecules in the ER; the Lmp proteins are components of the proteasome which cleave proteins specifically for MHC class I presentation (Janeway et al., 1999).
(2'-5')-Oligoadenylate Synthetase / dsRNA Dependent Protein Kinase
These are the two best-known IFN-b-induced proteins (Biron, 1998). (2'-5')-oligoadenylate synthetase polymerizes ATP in a unique 2'-5' fashion (Janeway et al., 1999); the resultant oligomers activate RNase L, which cleaves mRNA (Biron, 1998). dsRNA dependent protein kinase phosphorylates and inactivates elF2, a transcriptional initiator. Both (2'-5')-oligoadenylate synthetase and dsRNA dependent protein kinase act only in the presence of dsRNA, i.e. in virally infected cells. The net result of the action of these two proteins is to inhibit protein translation, which will retard viral replication (Biron, 1998).
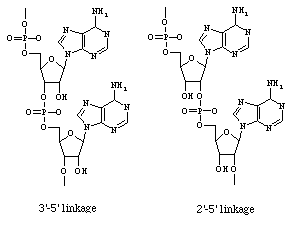
Figure 2. Alternate placement of
phosphodiester bond in polyribonucleotides. Left molecule:
normal 3'-5' linked RNA; right molecule: 2'-5' linked adenosine
oligomer produced by (2'-5')-oligoadenylate synthetase.
NK cell Activation
IFN-b is known to both activate and induce some proliferation in natural killer (NK) cells (Janeway et al., 1999). IFNs themselves are not mitogens, however, and this proliferation is probably caused by an intermediary cytokine which is induced by IFN-b (Biron, 1998). NK cells can kill cells which exhibit atypical patterns of MHC class I expression; such cells are generally virally infected (Janeway et al., 1999).
Cell Redistribution
The action of IFN-b has been implicated in observed changes in cell distribution, such as decreases in thoracic duct drainage and movement of nucleated spleen cells from red pulp to white pulp. Although the importance of these changes is not well established, it is hypothesized that such redistributions could be important in antigen presentation to T and B cells (Biron, 1998).
Rescue of T cells from Apoptosis
At the end of a successfully defeated infection, T cells die en masse by apoptosis as the immune system returns to a homeostatic balance. In order to preserve immunological memory, however, some T cells must avoid apoptosis and enter a G0/G1 memory state. These memory T cells are rescued from apoptosis by interacting with stromal cells, which secrete IFN-b and some IFN-a (Pilling et al., 1999). T cell apoptosis may be induced by either cytokine deprivation or ligation of Fas on the cell surface, but IFN-b is able to block both apoptotic pathways. The former apoptotic pathway is blocked by IFN-b-dependent upregulation of Bcl-x, an apoptotic inhibitor. Fas ligation-induced apoptosis occurs much too quickly to be blocked by upregulation of a gene, so IFN-b must block that apoptotic pathway by different means (Scheel-Toellner et al.,1999). The existence of a second blocking mechanism is supported by the results of Marrack et al. (1999), who found that IFN-b prevented T cell apoptosis without increased production of Bcl-x.
T Cell Cytotoxicity
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is a membrane-bound protein found on the surface of CD8 cytotoxic T cells. TRAIL is known to be involved in the anti-tumor action of CD8 cells, but TRAIL expression is also upregulated by type I IFNs (such as IFN-b) and may also be involved in antiviral T cell cytotoxicity (Kayagaki et al., 1999).
Other Genes Regulated by IFN-b
Der et al. (1998) found that IFN-b increased transcription
of well over 100 proteins in human fibrosarcoma cells. Induced
proteins ranged in function from cytochromes and cell scaffolding
proteins to immunologically active proteins such as Complement
components and dsRNA adenosine deaminase. These results
indicate that IFN-b has truly pleiotropic effects, many of which are not
fully understood.
Clinical Applications of IFN-b
Much clinical research on IFN-b is currently focused on its use as a treatment for multiple sclerosis (MS). MS is an autoimmune disease in which T cells mount an immune response against self myelin antigens in the glial cells of the central nervous system (Goodkin, 1999). In 1993, the FDA approved subcutaneous injections of IFN-b1b for treatment of MS (Revelle, 1993). IFN-b1b is a non-glycosylated form of IFN-b produced by E. coli (Arduini et al., 1999); it is marketed as Betaseron. Currently, IFN-b1a (a eukaryotic, glycosylated form obtained from hamsters) is also availabe under the tradename Avonex (Goodkin, 1999).
IFN-b treatment may ameliorate autoimmune attacks by restoring suppressor T cell function; cotreatment with all-trans-retinoic acid seems to increase this restorative action for unknown reasons (Qu et al., 1998). IFN-b may also inhibit the induction of inducible nitric oxide synthase (INOS) expression by IL-1 and IFN-g. Production of nitric oxide by INOS in astrocytes has been implicated as a factor in the parthenogenesis of MS (Hua et al., 1998).
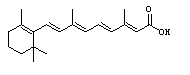
Figure 3. All-trans-retinoic
acid. Structure retrieved from Chemfinder.com
Although MS treatment is currently the most
prevalent application of IFN-b, researchers are also beginning to explore the use
of type I IFNs in treatment of hepatitis. Specifically,
clinical trials are currently underway for IFN-b1a in hepatitis C patients
(American Liver Foundation, 1997).
IFN-b Malfunctions
A search of recent immunological literature does not reveal any current research on IFN-b deficient organisms or IFN-b null mutations. It is possible that because of the pleiotropic effects of IFN-b, organisms lacking that protein would have multiple, overlapping immune deficiencies which would negate the usefulness of IFN-b- organisms as a research tool.
However, some recent research indicates that
IFN-b
may play a role in chronic inflammatory diseases such as rheumatoid
arthritis and atopic eczema. The chronic inflammatory response
in these diseases is known to be influenced by stromal cell-mediated
persistence of T lymphocytes in the areas of inflammation.
As described above, stromal cells rescue
T cells from apoptosis by secreting IFN-b. It is possible,
therefore, that inappropriate production of IFN-b may be responsible for
localized persistance of T cells and the resultant chronic inflammation
(Pilling et al., 1999).
Literature Cited
American Liver Foundation. 1997. Experimental treatment for hepatitis B and C. <http://gi.ucsf.edu/alf/alf/hepbcsf.html> Accessed 2 March 2000.
Arduini RM, Strauch KL, Runkel LA, Carlson MM, Hronowski X, Foley SF, Young CN, Cheng W, Hochman PS, Baker DP. 1999. Characterization of a soluble ternary complex formed between human interferon-b-1a and its receptor chains. Protein Science 8: 1867-1877.
Biron C. 1998. Role of early cytokines, including alpha and beta interferons (IFN-a/b), in innate and adaptive immune responses to viral infections. Seminars in Immunology 10: 383-390.
Chen Y, Wu W, Yang J, Sui S, Sun J, Dierich MP. 1999. Antibodies against human IFN-a and -b recognized the immunosuppressive domain of HIV-1 gp41 and inhibit gp41-binding to the putative cellular receptor protein p45. Immunology Letters 69: 253-257.
Der SD, Zhou A, Williams BRG, Silverman RH. 1998. Identification of genes differentially regulated by interferons a, b, or g using oligonucleotide arrays. Proceedings of the National Academy of Sciences, USA 95: 15623-15628.
Goodkin DE. 1999. Multiple sclerosis: Treatment options for patients with relapsing-remitting and secondary progressive multiple sclerosis. <http://www.msnews.org/goodkin1_99.htm> Accessed 2 March 2000.
Harada H, Taniguchi T, Tanaka N. 1998. The role of interferon regulatory factors in the interferon system and cell growth control. Biochimie 80: 641-650.
Hua LL, Lin JS, Brosnan CF, Lee SC. 1998. Beta inteferon prevents nitric oxide/peroxynitrate from damaging the central nervous system. <http://members.tripod.com/~ThJuland/nitric-oxide_beta.html>
Janeway CA, Travers P, Walport M, Capra JD. 1999. Immunobiology: The immune system in health and disease. 4th ed. New York: Elsevier Science/Garland Publishing. pp 385-386.
Kayagaki N, Yamaguchi N, Nakayama M, Eto H, Okumura K, Yagita H. 1999. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. Journal of Experimental Medicine 189: 1451-1460.
Marrack P, Kappler J, Mitchell T. 1999. Type I interferons keep activated T cells alive. Journal of Experimental Medicine 189: 521-529.
Min W, Pober JS, Johnson DR. 1998. Inteferon induction of TAP1: The phosphatase SHP-1 regulates crossover between the IFN-a/b and the IFN-g signal-transduction pathways. Circulation Research 83: 815-823.
Pilling D, Akbar AN, Girdlestone J, Orteu CH, Borthwick NJ, Amft N, Scheel-Toellner D, Buckley CD, Salmon M. 1999. Interferon-b mediates stromal cell rescue of T cells from apoptosis. European Journal of Immunology 29: 1041-1050.
Pitha PM, Au WC, Lowther W, Juang YT, Schafer SL, Burysek L, Hiscott J, Moore PA. 1998. Role of the interferon regulatory factors (IRFs) in virus-mediated signaling and regulation of cell growth. Biochimie 80: 651-658.
Qu ZX, Dayal A, Jensen MA, Arnason BG. 1998. All-trans retinoic acid potentiates the ability of interferon beta-1b. <http://members.tripod.com/~ThJuland/ra-beta1b.html> Accessed 2 March 2000.
Revelle M. 1993 07 Sept. FDA licenses interferon beta-1b. <http://www.fda.gov/bbs/topics/NEWS/NEW00424.html> Accessed 2 March 2000.
Russell-Harde D, Wagner TC, Perez HD, Croze E. 1999. Formation of a uniquely stable type I interferon receptor complex by interferon b is dependent upon particular interactions between interferon b and its receptor and independent of tyrosine phosphorylation. Biochemical and Biophysical Research Communications 255: 539-544.
Scheel-Toellner D, Pilling D, Akbar AN, Hardie D, Lombardi G, Salmon M, Lord JM. 1999. Inhibition of T cell apoptosis by IFN-b rapidly reverses nuclear translocation of protein kinase C-d. European Journal of Immunology 29: 2603-2612.
Return to Will White's homepage
![]()
For questions or comments, please contact wiwhite@davidson.edu.
Copyright 2000, Davidson College Department of Biology, Davidson,
NC 28036.