This web page was produced as an assignment for an undergraduate
course at Davidson College.
TNF-Alpha
Figure 1
Model of Murine TNF-Alpha at 1.4 A Resolution.
(Baeyens et al., 1998a). From NCBI
Structure Databank.
SYNONYMS
Lymphotoxin B, Cachectin.
FUNCTION
Tumor necrosis factor-alpha (TNF-A) is a pleiotropic inflammatory cytokine.
It was first isolated by Carswell et al. in 1975 in an attempt to identify
tumor necrosis factors responsible for necrosis of the sarcoma Meth A (Carswell
et al., 1975). Most organs of the body appear to be affected by TNF-A,
and the cytokine serves a variety of functions, many of which are not yet
fully understood. The cytokine possesses both growth stimulating
properties and growth inhibitory processes, and it appears to have self
regulatory properties as well. For instance, TNF-A induces neutrophil
proliferation during inflammation, but it also induces neutrophil apoptosis
upon binding to the TNF-R55 receptor (Murray, et al., 1997). The
cytokine is produced by several types of cells, but especially by macrophage.
Tracey and Cerami suggest two beneficial functions of TNF-A which have
lead to its continued expression (1990). First, the low levels of
the cytokine may aid in maintaining homeostasis by regulating the body's
circadian rhythm. Furthermore, low levels of TNF-A promote the remodeling
or replacement of injured and senescent tissue by stimulating fibroblast
growth.
Additional beneficial functions of TNF-A include its role in the immune
response to bacterial, and certain fungal, viral, and parasitic invasions
as well as its role in the necrosis of specific tumors. Lastly it
acts as a key mediary in the local inflammatory immune response.
TNF-A is an acute phase protein which initiates a cascade of cytokines
and increases vascular permeability, thereby recruiting macrophage and
neutrophils to a site of infection. TNF-A secreted by the macrophage
causes blood clotting which serves to contain the infection. Without
TNF-A, mice infected with gram negative bacteria experience septic shock
(Janeway et al., 1999).
The pathological activities of TNF-A have attracted much attention.
For instance, although TNF-A causes necrosis of some types of tumors, it
promotes the growth of other types of tumor cells. High levels of
TNF-A correlate with increased risk of mortality (Rink & Kirchner,
1996). TNF-A participates in both inflammatory disorders of inflammatory
and non inflammatory origin (Strieter al., 1993). Originally
sepsis was believed to result directly from the invading bacteria itself,
but it was later recognized that host system proteins, such as TNF-A induced
sepsis in response. Exogenous and endogenous factors from bacteria, viruses,
and parasites stimulate production of TNF-A and other cytokines. Lipopolysaccharide
from from bacteria cell walls is an especially potent stimulus for TNF-A
synthesis (Tracey and Cerami, 1993). When cytokine production increases
to such an extent that it escapes the local infection, or when infection
enters the bloodstream, sepsis ensues. Systematic edema results in
low blood volume, hypoproteinanemia, neutropenia and then neutrophilia
(Janeway et al., 1999). Body organs fail and death may result. Victims
of septic shock experience fever, falling blood pressure, myocardial suppression,
dehydration, acute renal failure and then respiratory arrest (Tracey and
Cerami, 1993).
TNF-A exhibits chronic effects as well as resulting in acute pathologies.
To link to a diagram of cellular responses involved in chronic inflammation,
please click here.
[Continue scrolling through the slides to view illustrations of chronic
inflammation]. If TNF-A remains in the body for a long time, it loses
its anti tumor activity. This can occur due to polymerization of
the cytokine, shedding of TNF receptors by tumor cells, excessive production
of anti-TNF antibodies, found in patients with carcinomas or chronic infection,
and disruptions in the alpha-2 makroglobulin proteinase system which may
deregulate cytokines. Prolonged overproduction of TNF-A also results
in a condition known as cachexia, which is characterized by anorexia, net
catabolism, weight loss and anemia and which occurs in illnesses such as
cancer and AIDS. Cachectin and TNF-A were once considered different
proteins, but in 1985 researchers discovered that the two proteins were
homologous (Beutler et al., 1985a).
Systematic Effects of TNF in Acute v.
Chronic Exposure
| ACUTE, HIGH DOSE |
CHRONIC, LOW DOSE |
| Shock and tissue injury |
Weight loss |
| Catabolic hormone release |
Anorexia |
| Vascular leakage syndrome |
Protein catabolism |
| Adult respiratory distress disorder |
Lipid depletion |
| Gastrointestinal necrosis |
Hepatosplenomegaly |
| Acute renal tube necrosis |
Subendocardial inflammation |
| Adrenal hemorrhage |
Insulin reisistance |
| Decreased muscle membrane potentials |
Enhanced rate of tumor metastic |
| Disseminated intravascular coagulation |
Acute phase protein release |
| Fever |
Endothelial activation |
Source : Tracey and Cerami 1994.
STRUCTURE/BINDING SITES
TNF-A is a trimeric protein encoded within the major histocompatibility
complex. It was first identified in its 17 kd secreted form, but
further research then showed that a noncleaved 27kd precursor form also
existed in transmembrane form (Perez, et al., 1990). Stimulated macrophage
produce 27kd TNF-A, which can either bind directly to TNFR-55 and TNFR-75
receptors through cell-to-cell contact or undergo cleavage and bind in
its soluble form. Due to its jelly roll like structure, which it
shares in common with viral coat proteins, it has been hypothesized that
TNF-A and viral originated from a common ancestor cell (Jones et al., 1989).
TNF-A shares only 36% amino acid sequence homology with TNF-B,
also called lymphotoxin (LT) (Meager, 1991). Yet, the tertiary structures
of the two proteins are remarkably similar and both bind to TNF receptors
TNFR-55 and TNFR-75. These receptors are expressed on all somatic
cells.
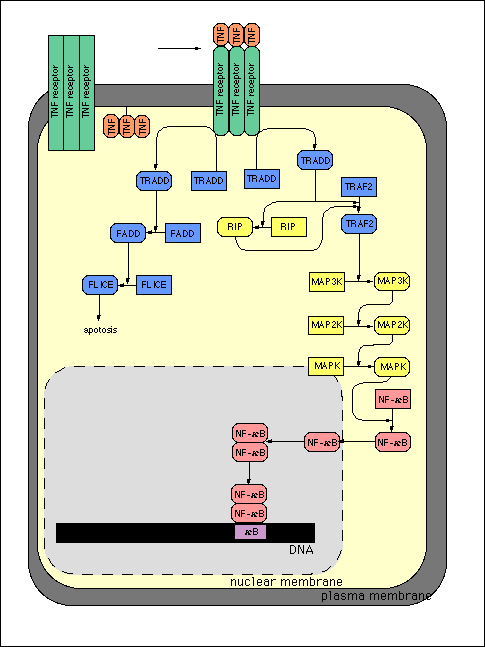
Figure 2
Signal transduction pathway initiated by trimeric TNF-alpha
binding to its receptor, TNFR to initiate receptor clustering
and signal transduction. (Higashi, 1998). Reproduced with permission
from author.
From Kyushu University Molecular Gene Technics Signaling
Pathway Database
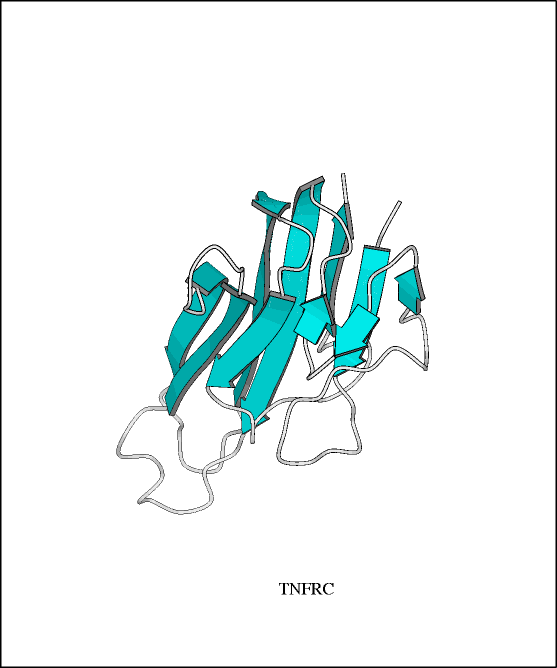
Figure 3
Model of TNFR-55, one of the two receptors
for TNF-A. (Pastore 1996). From the European Molecular
Biology Laboratory-The Bork Group Extracellular
Proteins
Database. Reproduced with permission
of author.
In addition to the transmembrane and soluble forms of TNF-A which bond
to the TNFR, TNF-A can penetrate cell membranes and form ion channels across
the membrane via acid induced transition from a hydrophilic to a
hydrophobic conformation. Researchers speculate that the viral protein
coat-like jelly role motif may facilitate membrane penetration (Kagan et
al., 1992).
TNF-A AND CLINICAL APPLICATIONS
TNF-A seems to serve as a mediator in various pathologies. A
few such examples include: Septic shock, Cancer, AIDS, Transplantation
rejection, Multiple Sclerosis, Diabetes, Rheumatoid arthritis, Trauma,
Malaria, Meningitis, Ischemia-Reperfusion Injury, and Adult respiratory
distress syndrome.
Since TNF-A plays a role in several diseases, a substantial amount of
research has been conducted concerning TNF-A therapies and anti-TNF-A therapies.
Because TNF-A exhibits anti tumor activity, research has been conducted
to determine the protein's effectiveness against certain forms of cancers.
Utilizing TNF-A tumoricidal activities has proved problematic, especially
due to the cytotoxin's systematic toxicity. While higher doses of
TNF-A may exhibit higher cytotoxicity, high doses also lead to systematic
toxicity (National Cancer Institute, 1995). Some studies involving
TNFR-75 and TNFR-55 mutants have suggested that the TNFR-75 receptor plays
a role in systematic toxicity, while TNFR-75 mutants will exhibit cytotoxicity
but not systematic toxicity (Van Ostade et al., 1993). Additionally,
a mutant form of TNF-A which exists only in the transmembrane form acts
only by cell-to-cell contact and may result only in cytotoxicity (Perez
et al.,1990), suggesting that mutant forms of TNF-A might effectively be
used therapeutically as against specific types of cancers.
Other research has focused upon inhibiting the effects of TNF-A in such
diseases as Rheumatoid Arthritis, Crohn's Disease, AIDS, bacterial septic
shock (caused by certain gram negative bacteria), and bacterial toxic shock
(caused by superantigens) as well as in prevention of alloreactivity and
graft rejection. Mutant mice that lack TNF-A are resistant to gram-negative
bacteria induced sepsis (Janeway et al. 1999), and anti-TNF monoclonal
antibodies have been used to effectively reduce or inhibit TNF-A activity
(Beutler et al., 1985b). One hypothetical advantage of treatment
with anti-TNF-A antibodies results from its role in multiple types of inflammation.
It is often difficult to determine that inflammation in burn and trauma
victims are of infectious etiology and warrant treatment with antibiotics;
therefore another treatment strategy might involve anti-TNF-A therapy (Strieter,
et al., 1993). Strategies for preventing TNF-A activity include
neutralization of the cytokine via either anti-TNF antibodies, soluble
receptors, or receptor fusion proteins; supression of TNF-A synthesis via
drugs such as cyclosporine A, glucocorticoides, or cytokine IL-10;
reduction of responsiveness to TNF-A via repeated low dose stimulation;
and lastly, by inhibition of secondary mediators such as IL-1, IL-6, or
nitric oxide (Tracey et al., 1993). Pharmaceutical companies
such as Peptech Limited have
developed different antibodies
to TNF-A, some of which inhibit various TNF-A functions and others
which do not affect protein activity. For instance, Remicade
(TM) is a chimeric Igk monoclonal anti-TNF antibody manufactured
by Centocor which has been
used to treat Crohn's disease--a chronic inflammatory disease of the intestines
(Centocor 2000). Soluble TNF-R will also neutralize TNF-A before
it can bind to its target cell receptor. Another drug, Enbrel
(TM) , developed by Immunex Corporation,
is a fusion of two soluble TNF receptors and a human immunoglobulin (Immunex
Corporation, Nov. 1999). It has been approved for treatment of rheumatoid
arthritis. Additionally, Chloroquine inhibits transcription of the protein
in macrophage (Zhu et al. 1993).
However, the efficacy of preventing septic shock has been questioned
as a result of recent research which suggests that, in the absence of TNF-A,
other cytokines will eventually initiate the inflammatory response.
The authors of this study speculate that TNF-A production may instead play
a key kinetic role by amplifying release of cytokines IL-A, IL-B, and IL-6
and thereby affecting the severity of a response to LPS (Amiot et al.,
1997). Additionally, eliminating the stimulatory affects of TNF-A
in diseases such as AIDS presents problems because inactivation of TNF-A
leaves the host at even greater risk for bacterial infections normally
countered by TNF-A activity.
References
Amiot, F., Fitting, C., Tracey, K. 1997. Lipopolysaccharide-Induced
Cytokine Cascade and Lethality in LT-A/TNF-A Deficient Mice. Molecular
Medicine. 3 (12) : 864-875.
Baeyens, K., De Bondt, H., Raeymaekers, A., Fiers, W., De Ranter, C.
1998a October 12. MMDB Structure Summary. <http://www.ncbi.nlm.nih.gov:80/cgi-bin/Entrez/Structure/mmdbsrv?form=6&db=t&Dopt=s&uid=11413>
Accessed 2000 February 17.
Baeyens, K., De Bondt, H., Raeymaekers, A., Fiers, W., De Ranter, C.
1998b October 12. Protein Databank Structure Explorer.
<http://www.rcsb.org/pdb/cgi/explore.cgi?job=graphics&pdbId=2TNF&page=&pid=>
Accessed 2000 February 25.
Beutler, B., Greenwald, D., Hulmes, J., Chang, M., Pan, Y., Mathison,
J., Ulevitch, R., Cerami, A. 1985a. Identity of Tumour Necrosis
Factor and the Macrophage-Secreted Factor Cachectin. Nature. 316
(8) : 552-554.
Beutler, B., Milsark, L., Cerami, A. 1985b. Passive Immunization
Against Cachectin/Tumor Necrosis Factor Protects Mice from Lethal Effects
of Endotoxin. Science 229 : 867-871.
Carswell, E., Old, L., Kassel, R., Green, N., Fiore, N., Williamson,
B. 1975. An Endotoxin-Induced Serum Factor that Causes Necrosis
of Tumors. Proceedings of the National Academy of Science.
72 (9) : 3666-3670.
Centocor. Centorcor Websit Home Page. <http://www.centocor.com/>
Accessed 2000 March 29.
Centocor. 2000 March 29. Prescribing Information for Remicade
(TM). <http:www.centocor.com/remicade-99.htm>
Accessed 2000 March 2.
Higashi, H. 1998 October13. Signal Pathway Mediated by TNF-alpha.
<http://www.grt.kyushu-u.ac.jp/spad/pathway/tnf.html>
Accessed 2000 February 25.
Immunex Corporation. 1999. Immunex Corporation. <http://www.immunex.com/>
Accessed 2000 February 29.
Immunex Corporation. 1999 November 23. Embrel Information.
<http://www.immunex.com/PRODUCTS/HTML/enbrelmardefault.html>
Accessed, 2000 February 29.
Janeway, c., Travers, P., Walport, M., Capra, J. Immunobiology
: The Immune System in Health and Disease. New York, N.Y :
Garland Publishers. 1999.
Jones, E., Stuart, D., Walker, N. 1989. Structure of Tumour
Necrosis Factor. Nature. 338 : 225-228.
Kagan, B., Baldwin, R., Munoz, D., Wisnieski, J. 1992. Formation
of Ion-Permeable Channels by Tumor Necrosis Factor-Alpha. Science.
255. 1427-1430.
Klatt, E. 1994. Webpath. Version 4.0 (1994-1999, 2000).
Inflammation. Chronic Inflammation, Diagram. <http://www-medlib.med.utah.edu/WebPath/INFLHTML/INFL066.html>
Accessed 2000 February 25.
Meager, Anthony. Cytokines. Englewood Cliffs, N.J. : Prentice
Hall. 1991.
Mucos. 1998 August 4. Additional Information : Tumor Necrosis
Factor. <http://www.mucos.de/uk/wiss/zytoktnf.htm>
Accessed 2000 February 26.
Murray, J., Barbara, J., Dunkley, S., Lopez, A., Van Ostade, X., Condliffe,
I., Haslett, C., Chilvers, E. 1997. Regulation
of Neutrophil Apoptosis by Tumor Necrosis Factor-Alpha : Requirements
for TNF-R55 and TNF-R75 for Induction of Apoptosis In Vitro. Blood.
90
(7) : 2772-2783.
National Cancer Institute. 1995 August . Biological
Therapies : Using the Immunse System to Treat Cancer. <http://www.graylab.ac.uk/cancernet/600072.html>
Accessed 2000 February 18.
Pastore, A. 1996 April 10. 3-D Images of MODULES in Extracellular
Proteins. <http://www.Bork.EMBL-Heidelberg.DE/Modules/3d/1tnr.gif>
Accessed 2000 February 25.
Peptech Limited. 2000 March 1. Peptech Limited. <http://www.peptech.com/>
Accessed 2000 February 2 .
Peptech Limited. 2000 March 1. TNF Binding Monoclonal Antibodies.
Table 1 : Properties of Key Antibodies. <http://www.peptech.com/ncs-tnf-ant-table1.htm>
Accessed 2000 February 27 .
Perez, C., Albert, I., DeFay, K., Zachariades, N., Gooding, L., Michael,
K. 1990. A Nonsecretable Cell Surface Mutant of Tumor Necrosis
Factor (TNF) Kills by Cell-to Cell Contact. Cell. 63 :
251-258.
Rink, L., Kirchner, H. 1996. Recent Progress in the Tumor Necrosis
Factor-Alpha Field. International Archives of Allergy and Immunology.
111 (3) : 199-209.
Strieter, R., Kunkel, S., Bone, R. 1993. Role of Tumor Necrosis
Factor-Alpha in Disease States and Inflammation. Critical Care Medicine.
21 (10 Supplement) : S447-S463.
Tracey, K., Cerami, A. 1990. Metabolic Response to Cachectin/TNF.
Annals
of the New York Academy of Sciences : Vol. 587. Eds.
Boland, B, Cullinan, J., Kimball, C. New York : New York Academy
of Sciences. 325-330.
Tracey, K., Cerami, A. 1994. Tumor Necrosis Factor : A Pleiotropic
Cytokine and Therapeutic Target. Annual Review of Medicine.
45 : 491-503.
Van Ostade, X., Vandenabeele, P., Everaerdt, B., Loetscher, H., Gentz,
R., Brockhaus, M., Lesslauer, W., Tavernier, J., Brouckaert, P., Fiers,
W. Human TNF Mutants with Selective Activity on the P55 Receptor.
Nature. 361 : 266-269.
Zhu, X., Ertel, W., Ayala, A., Morrison, M., Perrin, M., Chaudry, I.
1993. Chloroquine Inhibits Macrophage Tumor Necrosis Factor-Alpha
mRNA Transcription. Immunology. 80 (122) : 122-126.
Davidson CollegeImmunology
Home Page.

Send comments, questions, and suggestions to: rawolf@davidson.edu
{kind=link}