![]()
MULTIPLE SCLEROSIS
![]()
(Please click on the blue hyperlinks to read more about a particular subject)
Multiple sclerosis
(MS) is a chronic, demyelinating disease of the central nervous system
(Bebo et al., 1999), which usually strikes between the ages of 20 and 40.
Symptoms of this disorder include impairment of vision, sensation, muscle
strength and coordination, and cognitive processes. Each patient is affected
differently by the disorder (Wells, 1997), and the physical and emotional
progression of MS are unpredictable (National, 2000).
The different types of MS have recently
been classified into four major categories. Relapsing-remitting (RR) MS
is characterized by clearly defined flare-ups, followed by either complete
recovery or recovery accompanied by slight loss of function. Primary-progressive
(PP) MS involves a slow, continuous progession of the disease with only
brief episodes of improvement. Secondary-progressive (SP) MS commences
as relapsing-remitting MS and eventually converts to primary-progressive
MS. Finally, progressive-relapsing (PR) MS is associated with a continuous
decline from onset, yet has acute relapses, either with or without recovery
back to the level of disease which existed prior to the relapse (Wells,
1997).
The Central Nervous System and the Role of Myelin
The nervous system is a complex of
two major subdivisions, the central
nervous system (CNS) and the peripheral
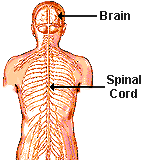
nervous system (PNS) (Waxman, 2000). The former, which is the target
of demyelination in multiple sclerosis, comprises the brain and spinal
cord, and depends upon the rapid and efficient transfer of nerve impulses,
primarily through action potentials, in order to properly control body
function (Wells, 1997) (Figure One).
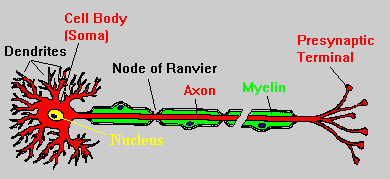
The axons of many nerves in the CNS
are covered by a myelin
sheath, which consists of multiple layers of lipid-rich membrane, produced
by oligodendrocytes
(Waxman, 2000). This sheath is divided into segments by short, unmyelinated
sections known as the nodes of Ranvier, and action potentials are able
to jump from node to node, dramatically increasing the speed of conduction
of a nerve impulse along the axon (Figure Two). This process, known as
saltatory
conduction, results from the abundance of sodium channels at the nodes,
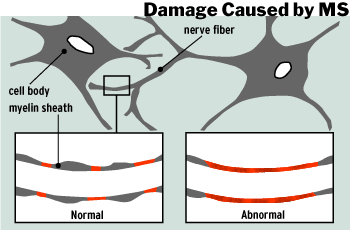
and the insulating properties of myelin. Multiple sclerosis is characterized
by the destruction of myelin sheaths in the CNS, thus slowing down or stopping
action potentials all together (Kalat, 1998).
Etiology
Although the precise etiology of MS is unknown, there are several major scientific theories concerning the source of the disease (Wells, 1997). It is now generally accepted that MS results from an autoimmune process, in which the immune system mounts an abnormal response against self antigens in the central nervous system (International, 1999). An MS exacerbation begins with the inflammation and swelling of the myelin, producing an area referred to as a lesion (Figure Three).
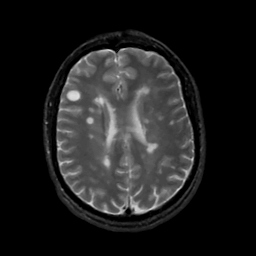
Figure Three. The large round
white spot in the
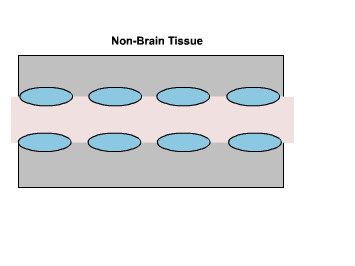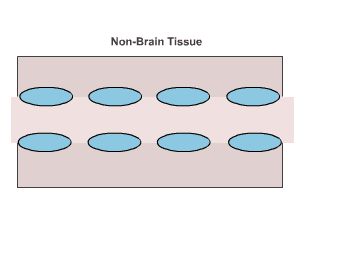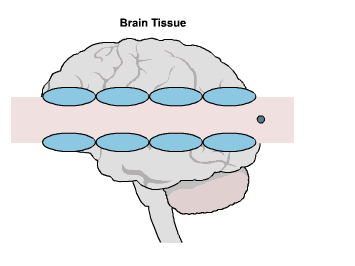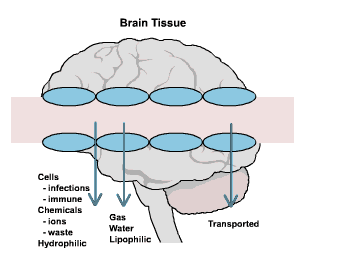
Though the exact cause of the autoimmune response is unknown, research indicates that a combination of several factors (International, 1999)- genetic, viral, and environmental- may be responsible (National, 2000). While MS is not a hereditary disease, having a first degree relative such as a parent or sibling, increases an individual's risk of developing MS. It is theorized that in these individuals, a genetic predisposition to react to environmental factors may exist. Upon exposure to the environmental agent, an autoimmune response can be triggered. A recent study by Bergsteinsdottir et al. (2000) suggests that certain genes may be shared by different autoimmune diseases, thus lending support to the theory of a genetic basis for MS (Bergsteinsdottir et al., 2000). It is also possible that a virus is the triggering factor in MS. Viruses are well-known causes of demyelination and inflammatory reactions (National, 2000) and may lie dormant in the body where they can eventually be activated and induce autoimmunity. It is likely that there is no single virus for MS, but that a common virus, such as measles, may trigger the disease (International, 1999). Patterns of migration and studies of epidemiology have shown that individuals who move from a high risk area of the world to a low risk area of the world before the age of fifteen, acquire the risk level associated with their new location. Thus, it appears that exposure to some environmental agent before puberty may predispose an individual to develop MS later in life (National, 2000).
B cells and autoantibodies
Colombo, et al. (2000) discuss two major etiological theories concerning MS. The first proposes that a viral or pathogenic infection precipitates the recruitment of inflammatory cells in the CNS, which may eventually lead to the onset of an autoimmune response. The second theory proposes that MS is triggered by a direct autoimmune response, which targets myelin antigen. Although research implicates T cells, one of the major infiltrates of MS, in the formation of inflammatory lesions, the production of autoantibodies has been shown to be an important factor in the demyelination of nerve cell axons. The involvement of B cells in MS has been investigated, and examinations of the cerebrospinal fluid (CSF) of MS patients reveals the presence of anti-myelin antibodies (Colombo et al., 2000). More specifically, these antibodies are often directed against myelin basic protein (MBP), although detection of anti-MBP antibodies is often regarded as a marker of neurological disease rather than its cause. A variety of other auto-antibodies have been identified in MS patients including anti-myelin-oligodendrocyte glycoprotein, anti-transaldolase, which binds to an enzyme expressed within oligodendrocytes, and antibodies to neoantigens. Neoantigens are expressed following infection of a cell by a virus, and are typically nonsense regions of normal DNA (Vincent et al., 1999). Using polymerase chain reaction (PCR) techniques, Colombo et al. (2000) were able to reveal an ongoing B cell response in MS patients to a relatively limited number of stimulating antigens, as there was an accumulation of clonally related B cells in the CSF of these patients.
T-cell mediated, Fas-dependent mechanisms
Sabelko-Downes et al. (1999) investigated another possible mechanism of myelin degeneration known as T-cell mediated, Fas-dependent death. This mechanism is an appealing one to consider, as it is not necessarily MHC-restricted (major histocompatibility complex-restricted) and the targets of CNS disruption do not express MHC. They propose that following initial stimulation, T cells which have been activated and have differentiated infiltrate the CNS. T cells specific for neuroantigen then encounter resident antigen-presenting cells (APC), which present antigen, and stimulate the T cells to secrete cytokines such as TNF-alpha and IFN-gamma. These cytokines initiate an inflammatory response, and CNS-specific, FasL+ T cells (stimulated by APCs expressing B7) begin to damage Fas+ targets such as oligodendrocytes. The damage produced in this early stage may perpetuate the inflammatory response and recruit additional T cells, B cells, and macrophages into the lesion, causing further damage. In addition to Fas-FasL dependent mechanisms, non-fasL-mediated factors such as the release of cytotoxic molecules may also damage oligodendrocytes and myelin (Sabelko-Downes et al, 1999).
Chemokines
Chemokines have also been implicated in the pathogenesis of MS during the progression of lesions. Staining for chemokines in the CNS of MS patients revealed low RANTES and MCP-1 expression associated with the endothelium in inflammation, which attract mononuclear cells from the blood across the blood-brain barrier and into the CNS. The local inflammatory response is maintained through the activation of resident glial cells and the expression of MCP-1, RANTES, and MIP-1alpha by astrocytes and MIP-1alpha, MIP-1beta, and MCP-1 by macrophages. Thus, inhibitory agents that block chemokine receptors may, in the future, provide treatment for MS (Woodroofe et al., 1999).
Experimental autoimmune encephalomyelitis (EAE) and the Beta7 Integrins
Although no single animal model currently exists, which imitates all of the features of MS, the prototypic model often utilized is experimental autoimmune encephalomyelitis (EAE). This disease can be induced in a number of species by immunization with components of CNS myelin, such as myelin basic protein, or CNS tissue (actively-induced EAE). In vitro, sensitized T cell lines can also be injected intravenously (adoptive- transfer EAE) (Gold et al., 2000). Kanwar et al. (2000) recently studied the involvement of beta7 integrins in the pathogenesis of EAE. These integrins are a subfamily of cell adhesion molecules, consisting of two members, alpha4beta7 and alphaEbeta7. By binding their ligands, MAdCAM-1, VCAM-1 and E-cadherin on the endothelial cells in the brain or on microvessels in the inflamed CNS, the beta7 integrins may be required to mediate the migration of leukocytes across the blood-brain barrier, producing chronic, non-remitting forms of EAE and, perhaps, MS. Thus, another source of treatment for MS may be the inhibition of certain adhesion pathways, particularly those involving the beta7 integrins (Kanwar et al., 2000).
Treatment
There is presently no cure for MS,
but a variety of treatments have been developed and are currently being
researched, which effectively act upon various facets of the disease (International,
1999).
Transforming growth factor- beta2
(TGF-beta2)
has recently been shown to reduce demyelination, the expression of viral
antigen, and the recruitment of macrophages in mice with Theilers murine
encephalomyelitis virus (TMEV), a demyelinating disease with pathology
similar to that observed in multiple sclerosis. TGF-beta2 is considered
to be an immunosuppressive cytokine based upon its ability to decrease
the proliferation of B and T cells and to suppress cytokine production.
In addition, studies have revealed that TGF-beta2 may also reduce NK cell
activity and the generation of cytotoxic T cells. Treatment with TGF-beta2
, three times weekly for thirty-five days following infection, resulted
in significantly smaller lesions and a decline in the number of antigen-positive
cells in the spinal cords of infected mice. Presumably, TGF-beta2 works
by decreasing the function of macrophages and their infiltration into the
CNS, thereby reducing the chronic demyelination characteristic of both
TMEV and MS (Drescher et al., 2000).
Interferon-beta
(IFN-beta) has been shown to be effective against relapsing-remitting MS
and clinical trials are currently underway to determine its effectiveness
in progressive MS (International, 1999). A recent study by Khademi et al.
(2000) revealed that IFN-beta, a cytokine itself, inhibits the effects
of proinflammatory cytokines such as interferon (IFN) gamma and tumor necrosis
factor (TNF) alpha. The reduction of IFN-gamma through upregulation of
IFN-beta in the CNS is potentially beneficial, as IFN-gamma is responsible
for the activation of macrophages and the promotion of TH1 immune responses
(Khademi et al., 2000). A number of types of IFN-beta are currently available.
The trade names for IFN-beta
1b are Betaseron and Betaferon and for IFN-beta 1a, are Avonex and
Rebif. The latter of these pharmaceutical drugs is still undergoing clinical
trials in the United States and Betaseron has been available as a prescribed
drug in the United States for 2 years. Around 20,000 people diagnosed with
MS are currently taking Betaseron in the United States (International,
1999).
Intravenous immunoglobulin (IVIg)
therapy is also showing promise in the treatment of MS. This form of treatment
involves the injection of antibodies derived from the blood of healthy
individuals into MS patients. (Wells, 1997). A recent study by Stangel
et al. (2000) indicates that IVIg may protect oligodendrocytes from damage
by antibody-mediated complement through antibody-binding and may also promote
myelin repair in damaged areas. Inhibition of inflammatory mechanisms,
rather than a direct effect on remyelinating cells as was previously thought,
may promote myelin repair (Stangel, 2000).
Treatment with 1,2-Dihydroxyvitamin
D3 may also be effective in resolving acute MS attacks in patients with
relapsing-remitting MS. Research reveals that 1,2-Dihydroxyvitamin D3,
a hormonally active vitamin D metabolite, significantly reduces the number
of macrophages accumulating in the inflamed CNS of mice with EAE (Nashold,
2000). In addition, in vitro, 1,25-Dihydroxyvitamin D3, inhibits the proliferation
of T cells and decreases the production of the cytokines, IL-2, IFN-gamma,
TNF-alpha (Cantorna, 2000).
Numerous other treatments are currently
available or being researched. To view a complete list please visit: http://www.msaa.com/msaa/litpro.htm.
A great deal of research is currently
underway to identify the exact mechanisms associated with the pathogenesis
of MS. While it may not be possible to improve all function lost to the
disorder, victims of MS should maintain their physical and mental condition
through rehabilitation and counselling programs. Research is not limited
to slowing or halting the progression of MS, as numerous studies are currently
investigating the possibility of repairing myelin, which could restore
lost function to victims of the disease (Wells, 1997). Early diagnosis
and commencement of treatment are also important and, thus, individuals
should discover, early on, the occurrence of MS within their own family.
Bebo, B., Adlard, K., Schuster, J., Unsicker, L., Vandenbark, A., & Offner, H. (1999). Gender Differences in Protection from EAE Induced by Oral Tolerance With a Peptide Analogue of MBP-Ac1-11. Journal of Neuroscience Research, 55, 432-440.
Bergsteinsdottir, K., Yang, H., Pettersson, U., & Holmdahl, R. (2000). Evidence for Common Autoimmune Disease Genes Controlling Onset, Severity, and Chronicity Based on Experimental Models for Multiple Sclerosis and Rheumatoid Arthritis. Journal of Immunology, 164, 1564-1568.
Cantorna, M. (2000). Vitamin D and Autoimmunity: Is Vitamin D Status an Environmental Factor Affecting Autoimmune Disease Prevalence? Proceedings of the Society for Experimental Biology and Medicine, 223, 230-233.
Colombo, M., Dono, M., Gazzola, P., Roncella, S., Valetto, A., Chiorazzi, N., Mancardi, G., & Ferrarini, M. (2000). Accumulation of Clonally Related B Lymphocytes in the Cerebrospinal Fluid of Multiple Sclerosis Patients. Journal of Immunology, 164, 5, 2782-2789.
Drescher, K., Murray, P., Lin, X., Carlino, J., & Rodriguez, M. (2000). TGF-Beta2 Reduces Demyelination, Virus Antigen Expression, and Macrophage Recruitment in a Viral Model of Multiple Sclerosis. Journal of Immunology, 164, 6, 3207-3213.
Gold, R., Hartung, H., Toyka, K. (2000). Animal models for autoimmune demyelinating disorders of the nervous system. Molecular Medicine Today, 6, 88-91.
International Federation of Multiple Sclerosis Societies (1999). The World of Multiple Sclerosis. Available: http://www.ifmss.org.uk/
Kalat, J. (1998). Biological Psychology, 6th ed. New York: Brooks/Cole Publishing Co.
Kanwar, J., Harrison, J., Wang, D., Leung, E., Mueller, W., Wagner, N., & Krissansen, G. (2000). Beta7 integrins contribute to demyelinating disease of the central nervous system. Journal of Neuroimmunology, 103, 146-152.
Khademi, M., Wallstrom, E., Andersson, M., Piehl, F., Di Marco, R., & Olsson, T. (2000). Reduction of both pro- and anti-inflammatory cytokines after 6 months of interferon beta-1a treatment of multiple sclerosis. Journal of Neuroimmunology, 103, 202-210.
Nashold, F., Miller, D., & Hayes, C. (2000). 1,25-Dihydroxyvitamin D3 treatment decreases macrophage accumulation in the CNS of mice with experimental autoimmune encephalomyelitis. Journal of Neuroimmunology, 103, 171-179.
National Multiple Sclerosis Society (2000) . MS Information. Available: http://www.nmss.org/
Sabelko-Downes, K., Russell, J., & Cross, A. (1999). Role of Fas-FasL interactions in the pathogenesis and regulation of autoimmune demyelinating disease. Journal of Neuroimmunology, 100, 42-52.
Stangel, M., Compston, A., & Scolding, N. (2000). Oligodendroglia are protected from antibody-mediated complement injury by normal immunoglobulins. Journal of Neuroimmunology, 103, 195-201.
Vincent, A., Lily, O., & Palace, J. Pathogenic autoantibodies to neuronal proteins in neurological disorders. Journal of Neuroimmunology, 100, 169-180.
Waxman, S. (2000). Correlative Neuroanatomy, 24th ed. New York: McGraw-Hill.
Wells, S. (1997). The Process and Medical Treatments. Multiple Sclerosis Association of America Online. Available: http://www.msaa.com/msaa/litpro.htm
Woodroofe, N., Cross, A., Harkness, K., Simpson,
J. (1999). The Role of Chemokines in the Pathogenesis of Multiple Sclerosis.
Advances in Experimental Medicine and Biology, 468, 135-150.

Link
back to Erin Woodall's Immunology Home Page
Link to Davidson College Department
of Biology
Link to Davidson College
Immunology Home Page

Please e-mail questions or comments to erwoodall@davidson.edu.