This web page was produced as an assignment for an undergraduate course at Davidson College.
Asthma is a very common disease of the airways that involves a plethora of factors. Asthma is characterized by airway hyperreactivity and mucus hypersecretion, two attributes that create chronic inflammation in the airways of asthmatics. Much of what is known so far about asthma is the significant role that the immune system plays. Most asthmatics are atopic, and thus are predisposed to allergies as a result of having a high sensitivity to many allergens due to high numbers of lymphocytes, eosinophils, and IgE. For these atopic individuals, asthmatic symptoms are the responses they have to the allergens that they are susceptible to. Yet, there are many nonatopic individuals who exhibit the same asthmatic symptoms as atopic individuals in responses to other factors such as exercise or infection. Why nonatopic and atopic asthmatics exhibit very similar symptoms in response to different triggers is not entirely clear, yet the pathophysiology of airway constriction in nonatopic asthmatics is very similar to that of atopic asthmatics (Renauld 2001). Much more is known about the important players in the immune system that trigger allergic asthma. Yet, the role of the immune system in allergic asthma is complex and difficult to pinpoint directly due to the role of a variety of cells and cytokines that contribute to the affliction of asthmatic symptoms.
Asthmatic symptoms and their triggers:
The most common allergen sources that asthmatics are especially sensitive to are grass pollens, animal danders, and house dust mite. When exposed to these allergen sources, asthmatic individuals have an allergic response that is concentrated in their airways. The hyperreactive nature of their airways, characterized by excessive smooth muscle contraction and inflammation in the airways, results in airway narrowing (Lambrecht 2006). Inflammation in the airways leads to airway narrowing because the tissue damage and subsequent airway remodeling performed by Th2 cells, eosinophils, neutrophils, mast cells, and other leukocytes thickens the airway wall. Airway narrowing accompanied by mucus hypersecretion contributes to the chronic symptoms of asthma such as wheezing, coughing, and sputum production (Renauld 2001).
Upon allergen exposure, asthma is triggered by the activation of mast cells in the submucosa of the lower airways. Mast cells constitutively bind to free IgE antibodies via their FcεRI receptors, receptors that are specific for the Fc portions of IgE antibodies. Cross-linking of IgE on mast cells occurs if bound by antigen that has multiple epitopes that are specific for the ideotypes on the variable portion of the IgE antibodies. Thus, an asthmatic individual must have had a primary exposure to the allergen in order to create memory B cells that synthesize and release IgE antibodies specific for the allergen. IgE cross-linking on mast cells leads to mast cell activation. Activated mast cells undergo degranulation and release histamine and other inflammatory factors into the extracellular space. These inflammatory factors aid in the recruitment of Th2 cells, eosinophils, and other leukocytes to the lungs (Janeway et al, 2005).
Lung epithelial cells also play a role in the recruitment of the important cellular players to the airways by producing the chemokine receptor CCR3 as well its ligands, CCL5 and eotaxin 1. CCR3 is also presented by eosinophils and T cells and thus are recruited to the inflamed airways when this chemokine receptor interacts with its ligands (Janeway et al, 2005).
Important players in allergic asthma:
Emerging research reveals that there are many factors that contribute to asthmatic symptoms in response to allergenic exposure. The mast cell, eosinophil, and Th2 cell are well-known major contributors to asthmatic symptoms, yet they are by no means the only cells involved. New research continues to reveal the complexity of allergic asthma and the components involved. A range of cells and cytokines have shown to be involved in the development of asthma and its symptoms.
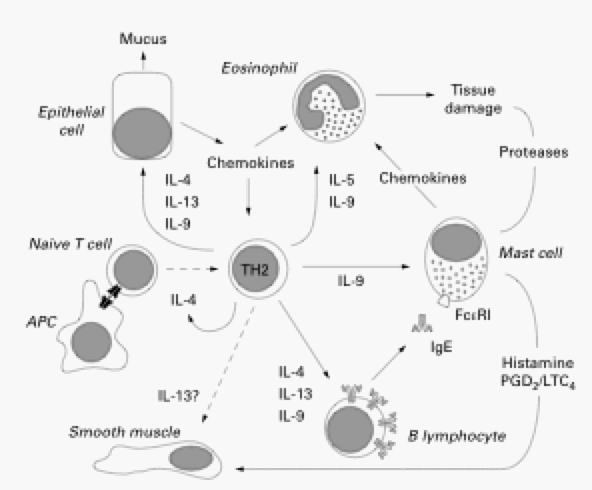
Figure 1. The cells involved in inducing symptoms of allergic asthma and the cytokines they secrete to activate other cells. APC, antigen presenting cell; PGD2, prostaglandin D2. (Permission pending. Renauld 2001)
Cells involved:
Th2 cells:
Th2 cells are a subset of CD4+ T cells that induce the effector mechanisms carried out by eosinophils and mast cells, and isotype-switching of B cells to produce IgE antibody. This subset of CD4+ T cells has a distinct cytokine secretion pattern that leads to further priming and activation of other Th2 cells and aids in the induction of effector mechanisms carried out by other cells (Janeway et al, 2005). Th2 cells are required for the development of airway eosinophilia (Renauld 2001) and secrete the cytokines interleukin-4 (IL-4), IL-13, IL-5, and IL-9. Th2 cell priming primarily relies on IL-4. The function of Th2 cells as well as the other cell players in allergic asthma can be best illustrated by outlining the functions of the cytokines that the important cell players secrete.
Cytokines involved:
IL-4
IL-4 carries out many functions such as the regulation isotype switching in B cells to IgE, induction of MHC II and CD23 expression on cell surfaces of antigen-presenting cells (APCs), induction of adhesion molecule expression on endothelial cells, chemokine production, and activation of mast cells and eosinophils (Karp 2000). The main role of IL-4 is carried out during the initial priming of Th2 cells and thus are necessary for the initial differentiation of Th2 cells (Herrick and Bottomly 2003). Once Th2 priming has occurred, IL-4 no longer plays a primary function in asthma induction. The overexpression of IL-4 leads to lymphocytic and eosinophilic inflammation, but without airway hyperreactivity (Renauld 2001).
IL-13
IL-4 was thought to be the key cytokine involved in allergic asthma until a redundancy of function was found in the cytokine IL-13. Asthmatics show increased mRNA expression in their sputum, indicating that IL-13 is involved in allergic asthma (Truyen et al, 2006). IL-13 binds to the α chain of the IL-4 receptor, shares many functions with IL-4, and its gene is located 25 kilobases upstream of the IL-4 gene. Yet, IL-13 is less involved in initial Th2 priming and more active in inducing the asthmatic symptoms that follow. IL-13 can induce allergic responses even in the absence of functional T cells (Karp 2000). IL-13 overexpression induces inflammation, mucus hypersecretion, subepithelial fibrosis, and eotaxin production. Eosinophil and IgE infiltration, and IL-5 production are only eliminated in the combined absence of both IL-4 and IL-13 (Renauld 2001).
IL-5
The increased expression of IL-5 mRNA in the sputum of asthma patients is a reflection of eosinophil infiltration in the lungs (Truyen et al, 2006). IL-5 induces an increase in eosinophil production in the bone marrow as well as the release of eosinophils from the bone marrow into circulation (Janeway et al, 2005).
IL-10
In contrast to the other discussed cytokines, IL-10 has inhibitory effects on the effector mechanisms that dominate in asthmatic and other allergic responses . IL-10 inhibits the production of pro-inflammatory cytokines and chemokines by macrophages, neutrophils, and eosinophils, expression of HLA-DR, and some co-stimulatory expression. IL-10 also has direct inhibitory effects on APCs and T cells and modulates eosinophil accumulation in airways by possibly inhibiting eosinophil production in bone marrow. Those with asthma have a decreased expression of IL-10 illustrating IL-10’s important role in regulating allergic responses (Yssel et al, 2001).
Eosinophils:
Eosinophils play a prominent role in initiating asthmatic symptoms by causing tissue damage in the airways of the lungs. Unlike mast cells, they do not constitutiveley express FcεRI on their surface. Once activated by their appropriate cytokines and chemokines such as CCL5 and eotaxins, they express FcεRI on their surfaces and bind to IgE (Janeway et al, 2005). When cross-linked by antigen, eosinophils release major basic protein, eosinophil cationic protein, and free radicals. These secreted products can initiate mast cell and basophil degranulation as well as cause significant tissue damage in the airways. Other inflammatory cells respond to the tissue damage by remodeling the airways. This leads to hyperplasia and hypertrophy of the smooth muscle layer and mucous glands, and thus narrowed airways (Renauld 2001). Eosinophils become part of a positive feedback loop by synthesizing prostaglandins, leukotrienes, and cytokines that can activate other eosinophils (Janeway et al, 2005). While the eosinophil contributes to asthmatic symptoms via tissue damage and airway remodeling, it is not required for allergen-induced airway hyperresponsiveness (Renauld 2001).
Mast cells:
As stated previously, mast cells contribute to the triggering of asthma via their FcεRI receptors that constitutively bind free IgE. When allergen cross-links surface IgE, the mast cell releases histamine, tryptases, chymase, heparin, and synthesizes lipid mediators such as leukotriene C4 and prostaglandin D2. All of these play roles in bronchoconstriction and the recruitment of inflammatory cells to the airways. Along with recruiting inflammatory cells, histamine also plays a role in the activation of inflammatory cells, dendritic cells, and T cells (Robinson 2004).
The mast cell’s involvement in recruiting inflammatory cells is not limited to asthma, but to other allergic responses as well. It has been demonstrated that mast cells also have an involvement that is specific to an asthmatic response by interacting with airway smooth muscle (ASM). ASM produce a variety of pro-inflammatory cytokines and chemokines that recruit and retain mast cells in the airways. One such cytokine that ASM produces is stem cell factor (SCF). SCF aids in the recruitment and survival of mast cells as well as in the differentiation of mast cell precursors to mature mast cells. Evidence of the role of mast cells in asthma is demonstrated by the presence of histamine, prostaglandin D2, and tryptase in the bronchoalveolar lavage fluid of asthmatics. The presence of these components suggest mast cell degranulation (Robinson 2004).
Natural killer T cells:
Until recently, Th2 cells were thought to be the major inducers of effector mechanisms that lead to the development of asthma. The cytokine that Th2 cells depend on for priming is IL-4. Th2 cells synthesize and release IL-4 on activation, yet the piece of the puzzle missing was the source of IL-4 before Th2 priming occurs. Research in the past few years has demonstrated the role of natural killer (NK) T cells in initiating the effector mechanisms carried out by Th2 cells and other leukocytes. NKT cells are T-cell receptor (TCR)+ CD1d-restricted cells that express CD4. They are given the name NKT cells because of the characteristics they share with NK cells and CD4 T cells (Meyer et al, 2005).
NKT cells express an invariant TCR that consists of Va24-Ja18 in humans. Unlike the classic CD4+ T cell, NKT cells do not have highly specific receptors that only recognize a specific MHC and peptide. NKT cells recognize bacterial endogenous glycolipid antigens presented by CD1d, an MHC class I-like protein. They recognize endogenous glycolipids, glycolipids found in pollens, bacterial glycospingolipids found in the Sphingomonadaceae and Rickettsiaceae, a self-glycolipid isoglobotrihexosyl ceramide (iGb3), and a synthetic glycolipid α-galactosyl ceramide (Meyer et al, 2005). When NKT cells encounter one of these glycolipids presented by CD1d, they respond with a similar cytokine profile to that of Th2 cells, and produce IL-4 and IL-13. There are two pathways by which NKT cells can produce IL-4 and IL-13. In one pathway, NKT cells can be activated by CD1d presenting glycolipids presented by an APC. In the other pathway, NKT cells activate Th2 cells to secrete IL-4 and IL-13. In this second pathway, dendritic cells produce CDL25, a ligand that binds to CCR9 on NKT cells, and this leads to phosphorylation on CD226 on NKT cells. This signaling pathway allows for NKT cells to activate Th2 cells via cell-to-cell contact (Kay 2006). Thus, NKT cells provide a link between innate and adaptive immunity by activating the Th2 cells of adaptive immunity. IL-4 boosts the priming of allergen-specific CD4+ Th2 cells and IL-13 aids in the development of asthmatic symptoms.
Akbari et. al demonstrated the ability of NKT cells to rapidly induce AHR, airway inflammation, and non-specific IgE production upon direct activation by glycolipid antigens. While NKT cells aid in adaptive immunity with the ability to activate Th2 cells, they can illicit asthmatic symptoms independent of eosinophils, B cells, and MHC-II restricted CD4+ Th2 cells, and thus of adaptive immunity. Yet, adaptive immunity may contribute to the activation of NKT cells. Inflammation and tissue damage induced by Th2 cells may uncover self-glycolipid antigens, such as IGb3, that can activate NKT cells. NKT cells constitute less than 1% of CD4+ T cells in the blood and constitute 63% of CD4+ T cells in patients with moderate-to-severe asthma, illustrating their prominent involvement in allergic asthma (Akbari et al, 2006).
Regulatory T cells:
Regulatory T cells are a subset of T cells that aid in the regulation of immune responses by inhibiting the activity of Th1 and Th2 cells. There are naturally occurring regulatory T cells, known as T-regs and three subsets of inducible regulatory T cells: Th3, TR-1, and NKT cells (Tournoy et al, 2006). TR1 cells are the subset of regulatory T cells most involved in allergic asthma (Akbari et al, 2003). TR1 cells exert immune regulation activity via cell-cell interactions and by secreting the immune suppressing cytokines IL-10 and TGF-β. The induction of antigen-specific tolerance involves the activation of TR1 by an APC and the subsequent suppression of Th1 or Th2 cells by TR1 cells. Not only can TR1 cells inhibit Th2 cell activation, they have been found to also reverse a Th2-dominated immune response. The role of regulatory T cells in suppressing the effector mechanisms that induce asthmatic symptoms indicate that asthmatics may lack functional regulatory T cells that can inhibit an asthmatic response (Tournoy et al, 1006).
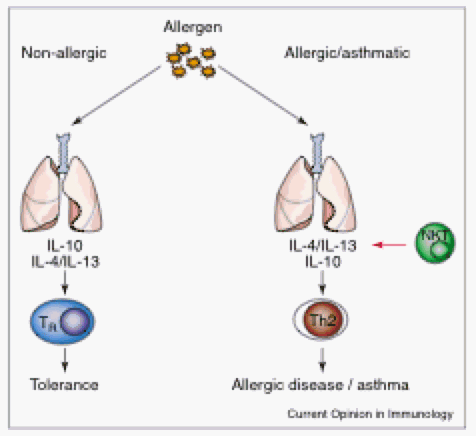
Figure 2. Allergen challenge on non-allergenic individuals vs. allergic asthmatic individuals results in different T-cell pathways. Non-allergenic individuals respond to allergen with IL-10 cytokines and T regulatory cells. Asthmatic individuals respond to allergen with Th2 cells and NKT cells and the prominent cytokines they that these cells secrete, IL-4 and IL-13. (Permission pending. Akbari et al, 2003)
Dendritic cells (DCs):
Dendritic cells are APCs that have a two-fold role in asthma due to the presence of two subsets of dendritic cells in the lungs. One subset of DCs, respiratory tract myeloid DCs (mDCs) aid in the development of asthma symptoms, while the other subset, plasmacytoid DCs (pDCs) aid in respiratory tolerance to allergens. mDCs are the prominent APC that activates CD4+ cells to produce and secrete cytokines. In conditions that lead to sensitization to inhaled antigens, mDCs outnumber pDCs, the regulatory subset of DCs (Lambrecht 2006). pDCs exhibit a regulatory role by either inhibiting mDC-driven T-cell activation or by inducing T regulatory cells. Either mechanism of function provides a form of protective immunity and respiratory tolerance (Tournoy et al, 2006). In conditions of respiratory tolerance, pDCs outnumber mDCs in the airways (Lambrecht 2006).
Proposed Treatments:
Influencing Th1/Th2 balance:
Since asthma and other allergic responses are the result of a Th2-dominated response, it was initially thought that resetting the cytokine balance to induce Th1 would counterbalance Th2 activity. A Th1-dominated response can be induced by administering the cytokines IFN-γ and IL-12 or infectious agents (Tournoy et al, 2006). While this method is efficient in suppressing eosinophilic airway inflammation, a Th1-dominated response does not have the desired effect on reducing allergic responses. A Th1-dominated response results in the recruitment of Th1 cells and enhancement rather than a reduction of airway inflammation (Akbari et al, 2003). Now, it is becoming realized that resetting the cytokine balance to regulatory T cells rather than to Th1 cells may result in a decrease in asthmatic symptoms.
Treatments:
Omalizumab:
Omalizumab is a humanized monoclonal antibody that blocks FcεRI, the Fc receptor that binds the Fc portion of IgE antibodies. By blocking this receptor, IgE is not able to bind to these receptors and thus cross-linking cannot be induced on the cells that express the FcεRI receptor. This treatment has shown to be very effective in inducing a decrease in IL-13 (Holgate et al, 2005), IL-4, eosinophils in epithelial and submucosal compartments, T cells, B cells, and other cells that stain as IL-4+ (Scheinfeld 2005).
Corticosteroids (glucocorticoids):
The use of inhaled and oral corticosteroids is a prominent treatment in reducing asthmatic symptoms. Corticosteroids reduce airway inflammation, airway hyperresponsiveness, allergen-induced peribronchial fibrosis (Miller et al, 2006), and Th2 cytokine synthesis (Xystrakis et al, 2006). Corticosteroids are able to induce their inhibitory effects by inducing regulatory gene expression and suppressing inflammatory gene expression. The mechanism in which corticosteroids exert their effects is by reversing histone acetylation of activated inflammatory genes through the binding of glucocorticoid receptors to coactivators. This prevents the transcription of inflammatory genes and activates glucocorticoid receptors. Activated glucocorticoid receptors bind to promoter regions of anti-inflammatory genes, such as IL-10, and activate their transcription (Barnes 2006).
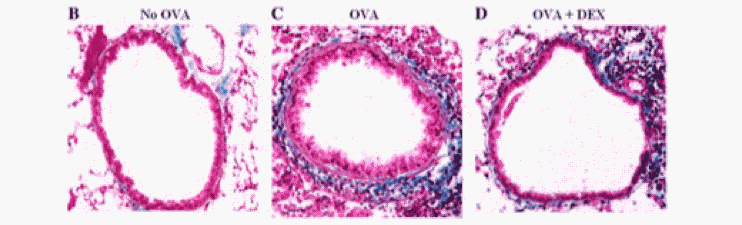
Figure 3. The effects of ovalbumin (OVA) challenge on airway remodeling in the airways. A lung section of non-OVA-challenged control mice with no airway remodeling (B). A lung section of mice challenged with OVA for 3 months and the subsequent airway remodeling (C). A lung section of mice challenged with OVA for 3 months and treated with corticosteroids (D). (Miller et al, 2006)
While corticosteroids are generally successful in reducing asthmatic symptoms, there are some who do not respond to corticosteroids and thus are known as steroid resistant. Steroid resistant asthmatics have CD4+ T cells that fail to induce IL-10 synthesis following stimulation of corticosteroids (Xystrakis et al, 2006). Steroid resistance may be the result of abnormalities in the glucocorticoid receptor signaling pathways (Barnes 2006). One method of combatting steroid resistance is oral administration of vitamin D3 to steroid-resistant patients. Vitamin D3 enhances these patients' response to glucocorticoids (Xystrakis et al, 2006).
References
Akbari, O., Faul, J.L., Hoyte, E.G., Berry, G.J., Wahlstrom, J., Kronenberg, M., DeKruyff, R.H., and Umetsu, D.T. CD4+ invariant T-cell-receptor+ natural killer T cells in bronchial asthma. N. Engl. J. Med. 354: 1117-1129, 2006.
Akbari, O., Stock, P., DeKruyff, R.H., and Umetsu, D.T. Role of regulatory T cells in allergy and asthma. Current Opinion in Immunology 15: 627-633, 2003.
Barnes, P.J. Corticosteroid effects on cell signaling. Eur. Respir. J. 27: 413-426, 2006.
Herrick, C. A. and Bottomly, K. To respond or not to respond: T cells in allergic asthma. Nature 3: 405-412. Review. 2003.
Herrick, C.A., MacLeod, H., Glusac, E., Tigelaar, R.E., and Bottomly, K. Th2 responses induced by epicutaneous or inhalational protein exposure are differentially dependent on IL-4. The Journal of Clinical Investigation 105: 765-775, 2000.
Holgate, S.T., Djukanovic R., Casale, T., and Bousquet J. Anti-immunoglobulin E treatment with omalizumab in allergic diseases: an update on anti-inflammatory activity and clinical efficacy. Clin. Exp. Allergy 35: 408-416, 2005.
Janeway, C.A., Travers, P., Walport, M., Shlomchik, M.J. 2005. Immunobiology. New York. Garland Science.
Karp, M.W., The gene encoding interleukin-13: a susceptibility locus for asthma and related traits. Respir. Res. 1: 19-23, 2000.
Kay, A.B. Natural killer T cells and asthma. N. Eng. J. Med. 11: 1186-1188, 2006.
Lambrecht, B.N. An unexpected role for the anaphylatoxin C5a receptor in allergic sensitization. The Journal of Clinical Investigation 116: 628-632, 2006.
Meyer, E.H., Goya, S. Akbari, O., Berry, G.J., Savage, P.B., Kronenberg, M., Nakayama, T., DeKruyff, R.H., and Umetsu, D.T. Glycolipid activation of invariant T cell receptor+ NK T cells is sufficient to induce airway hyperreactivity independent of conventional CD4+ T cells. PNAS 103: 2782-2787, 2006.
Miller, M., Cho, J.Y., McElwain, K., McElwain, S., Shim, J.Y., Manni, M., Baek, J.S., and Broide, D.H. Corticosteroids prevent myofibroblast accumulation and airway remodeling in mice. AJP-Lung 290: 162-169, 2006.
Renauld, J.C. New insights into the role of cytokines in asthma. J. Clin. Pathol. 54: 577-589, 2001.
Robinson, D.S. The role of the mast cell in asthma: Induction of airway hyperresponsiveness by interaction with smooth muscle? J. Allergy. Clin. Immunol. 114: 58-65, 2004.
Scheinfeld, N. Omalizumab: A recombinant humanized monoclonal IgE-blocking antibody. Dermatology Online Journal 11 (1): 2, 2005.
Tournoy, K.G., Hove, C.V., Grooten, J., Moerloose, K., Brusselle, G.G., and Joos, G.F. Animal models of allergen-induced tolerance in asthma: are T-regulatory-1 cells (Tr-1) the solution for T-helper-2 cells (Th-2) in asthma? Clin. Exp. Allergy 36: 8-20, 2006.
Truyen, E., Coteur, L., Dilissen, E., Overbergh, L., Dupont, L.J., Ceuppens J.L., and Bullens, D.M.A. Evaluation of airway inflammation by quantitative Th1/Th2 cytokine mRNA measurement in sputum of asthma patients. Thorax 61: 202-208, 2006.
Xystrakis, E., Kusumakar, S., Boswell, S., Peek, E., Urry, Z., Richards, D.F., Adikibi, T., Pridgeon, C., Dallman, M., Loke, T.K., Robinson, D.S., Barrat, F.J., O'Garra, A., Lavender, P., Lee, T.H., Corrigan, C., and Hawrylowicz, C.M. Reversing the defective induction of IL-10-secreting regulatory T cells in glucocorticoid-resistant asthma patients. J.Clin. Invest. 116: 146-155, 2006.
Acknowledgements:
Special thanks to Dr. Allen Dozor for guiding me in my research for the making of this page.
© Copyright 2006 Department of Biology, Davidson College, Davidson,
NC 28036
Send comments, questions, and suggestions to: kaacker@davidson.edu