This web page was produced as an assignment for an undergraduate course at Davidson College.
What Is Guillain-Barre Syndrome (GBS)?
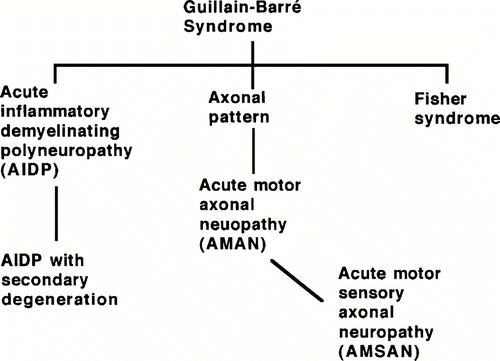
Guillain-Barre Syndrome (GBS) is a rare, post-infectious, inflammatory autoimmune disease that is characterized by an ascending limb weakness and numbness in the extremities that can progress, in some cases, to paralysis (Yuki, 2005). The annual incidence in the population ranges from 0.4-4.0 cases per population of 100,000. GBS can be divided into three subtypes that can be differentiated through electrodiagnostic techniques: acute inflammatory demyelinating polyradiculoneuropathy (AIDP), acute motor axonal neuropathy (AMAN), acute motor and sensory axonal neuropathy (AMSAN), and Miller Fisher syndrome (MFS) (Kuwabara, 2004).
Figure 1. Depiction of GBS subtypes and their relationships according to type of neuropathy within the GBS classification. (Image from Ho et al., 1998. Copyright 1998 by Annual Reviews, Inc. Permission pending.)
AIDP is the most common form of GBS in developed countries accounting for 90% of cases (Moran et al., 2005). GBS can occur at all ages and its development is usually associated with infections by bacteria and viruses. The mechanism proposed thus far for the development of GBS is molecular mimicry. In summary, the body mounts an antibody response against a pathogen protein that resembles neural glycolipids (gangliosides), resulting in an an attack on the peripheral nervous system. Most patients recover eventually with varying degrees of nerve damage. However there is an 8-10% mortality rate associated with GBS attributable to respiratory failure in advanced cases (Kuwabara, 2004). Treatments consist of immunoglobulin therapy or plasma exchange (along with intravenous corticosteroids in some cases) (Harel and Shoenfeld, 2005; Shahar, 2006).
Infection and Peripheral Nervous System Inflammation Response (Overview)
Infection
Most of the pathogens that are known to cause GBS gain entry to the body through mucosal or gut epithelium. They can enter the body by the use of multifenestrated cells or other mechanisms to cause an infection. The innate immune response results in the uptake of the pathogens by immature antigen presenting cells (APCs). After migration to lymph nodes, a mature, differentiated APC can present peptides in MHC class II molecules and activate CD4 T cells that recognize antigens from the infectious pathogen. B cells can be activated as well by newly activated Th2 cells. This produces a cell mediated and humoral response against the pathogen (Janeway, 2005). In the case of C. jejuni infection, antibodies will be produced, leading to activation of the complement system and phagocytosis of the bacteria. However, in rare cases the antibodies produced against certain C. jejuni antigens will also bind to gangliosides of the neurvous tissue causing complement activation and damage by phagocytes. This results in damage to peripheral nervous tissues, which leads to demyelination and axonal damage (Kuwabara, 2004).
Periheral Nervous System Inflammatory Response
The peripheral nervous system refers to the network of nervous tissue beyond the central nervous system (brain and spinal cord). The peripheral nerves are covered by a blood-nerve barrier that prevents the normal infiltration of macromolecules. However, the exception to this is that lymphcytes can move in and out of peripheral nervous tissue. This allows for immune protection of the macrophages and endothelial cells that reside in the tissue. In the case of GBS, which is an inflammatory disorder, autoantibodies are able to cross the blood-nerve barrier while in its normal state. The blood nerve barrier is made up of endothelial cells with tight junctions that can be modified under inflammatory conditions to allow passage of effector cells and macromolecules. However, when the autoantibodies cross the barrier and bind to gangliosides of neural tissue, they can activate complement cascades and the resident macrophages by their FcIII receptors inducing cytokine production and inflammation within the nerve tissue. It was also found that endoneural macrophages had the capacity to express CD1 (CD1a, b, and c), which allows them to present glycolipids such as gangliosides to some T cell subsets along with dendritic cells (Hughes et al., 2006). The inflammation due to cytokines and causes recruitment of leukocytes and damage to the nerve tissue by one of four mechanisms: CD8 T cell lysis, complement-mediated attack, cytokine and free radical damage via phagocytes, and antibody-mediated interference of nerve conduction (Ho et al., 1998). There are several places in the peripheral nervous system where the inflammation response of GBS can begin, depending on the subtype that develops and infectious pathogen. These locations are illustrated in Figure 2 and include: dorsal root ganglia, nodes of Ranvier, and schwann cell myelin (Ho et al., 1998).
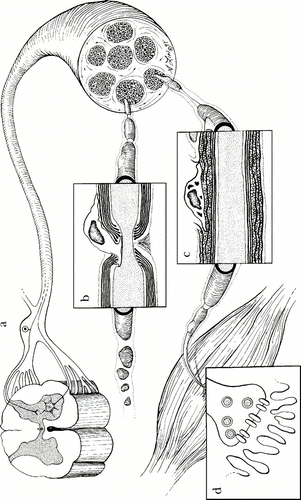
Figure 2 . Locations of GBS peripheral nerve attack in peripheral nervous system. a) Dorsal root ganglia can be the target of an antibody response in MFS. b) Nodes of Ravier are a target of immune response in AMAN. c) Schwann cell myelin surface proteins can be the target of antibody binding in AIDP. d) Neuromuscular junction (not known to be involved with GBS). (Image from Ho et al., 1998. Copyright 1998 by Annual Reviews, Inc. Permission pending.)
Pathogens and Autoimmunity in GBS Subtypes
Two-thirds of GBS cases are associated with prior acute infection of several bacterial species and virusus. Campylobacter jejuni, cytomegalovirus, Epstein-Barr virus, Mycoplasma pneumoniae, Haemophilus influenzae, and Varicella-zoster virus have been found in patient serum after the onset of GBS (Kuwabara, 2004). The most commonly proposed mechanism for the development of autoimmune disease is molecular mimicry (Yuki, 2005). Molecular mimicry refers to the situation where the pathogen and host share nearly identical antigens, which induces an antibody and T cell immune response that is cross reactive. There is more than one way that an immmune response can become cross-reactive. The pathogen and host can have homologous or identical amino acid sequences or the host B cell receptors and T cell receptors can recognize non-homologous peptides (Ang et al., 2004). The strongest evidence for the molecular mimicry hypothesis has come from discoveries in research with C. jejuni strains, the most common pathogen associated with GBS (specifically AMAN) (Neisser et al., 2000; Yuki, 2005). Since definite mechanisms and evidence of correlation between autoimmune responses and infection are limited in the cases of many pathogens listed above, only those with evidence-based correlations with GBS subtypes will be further described.
AIDP-associated Infection
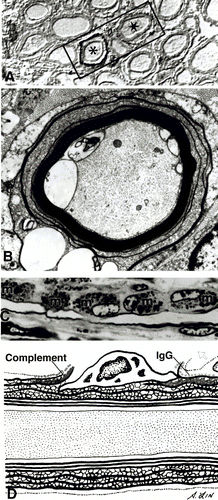
Figure 3. Immunopathology of AIDP. A) Immunostaining showing C3d surrounding two nerve fibers indicated by asterisks (*) B) Complement activation and influx of calcium along with demyelination. C) Complement-mediated activation of macrophages. D) Depiction of pathology in AIDP involving complement activation and macrophage-mediated demyelination. (Image from Ho et al., 1998. Copyright 1998 by Annual Reviews, Inc. Permission pending. )
Cytomegalovirus (CMV), a cause of respiratory tract infections, is the second most common pathogen linked to cases of GBS in Europe and Japan. Autoantibodies against the human ganglioside GM2 have been isolated in patients with a CMV infection and GBS symptoms. However, no definitive epitope mimics have been described despite the fact that CMV is correlated with so many cases of AIDP. Development of AIDP is seen predominantly in the cranial and sensory nerves as opposed to motor nerves. Also, the immune response elicited in AIDP is focused on the schwann cell or myelin sheath. Damage to the myelin or schwann cell results in demyelination that is characteristic of AIDP (Figure 3) (Kuwabara, 2004).
AMAN-associated Infection
Infection by C. jejuni, a cause of bacterial gastroenteritis, is the leading cause of AMAN worldwide (Kuwabara, 2004; Yuki, 2005). Studies show that the production of autoantibodies by C. jejuni infection occurs in only 1 out of 3285 patients with C. jejuni enteritis (Koga et al., 2006). Also, evidence of host susceptibility has been shown for patients with HLA B35 and HLA DQB 1*03, but because of conflicting data, the results are inconclusive (Kuwabara, 2004). It has been found that only certain strains of C. jejuni are associated with GBS/AMAN cases. The strains are divided by serotype based on their low molecular weight type lipopolysaccharide (LPS), called lipooligosaccharide (LOS). Serotypes most commonly associated with AMAN were HS:19 and HS:41. Also, a polymorphism in the gene cstII(Thr51) has been found to be closely associated with development of anti-GM1 and anti-GD1a autoantibodies (Koga et al., 2006).
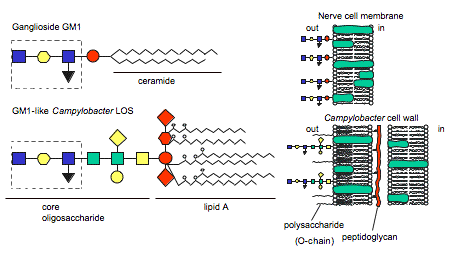
Figure 4 . Similarity between human ganglioside GM1 and GM1-like epitope of C. jejuni LOS. (Image from Ang et al., 2004. Reproduced with permission of the author.)
The hypothesis of molecular mimicry is based on the fact that the bacterial LOS induces IgG, IgA, and IgM auto anitibodies against human gangliosides due to LOS ganglioside-mimicking epitopes (Figure 4) (Moran et al., 2005). Autoantibodies have been isolated in GBS patients' serum and found to recognize C. jejuni LOS and human gangliosides GM1, GM1b, GD1a, and GalNAc-GD1a epitopes, providing evidence for molecular mimicry (Ang et al., 2004; Yuki, 2005). Furthermore, Moran et al. concluded that the IgG LOS-induced anti-GM1 antibodies bound to sites at the nodes of Ranvier in humans (Figure 5). This is important because other studies have concluded that antibodies bound to nodes of Ranvier disrupt Na+ and K+ channels, interferring with nerve conduction (2005).
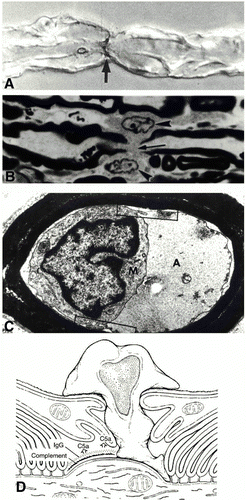
Figure 5 . Immunopathology of AMAN subtype of GBS. A) immunostaining of C3d at the node of Ranvier. B) Ventral root node of Ranvier lengthened along with macrophages. C) Periaxonal space with macrophage inside extending processes around the axon. D) Depiction of AMAN pathology involving antibodies, complement, and macrophage invasion. (Image from Ho et al., 1998. Copyright 1998 by Annual Reviews, Inc. Permission pending.)
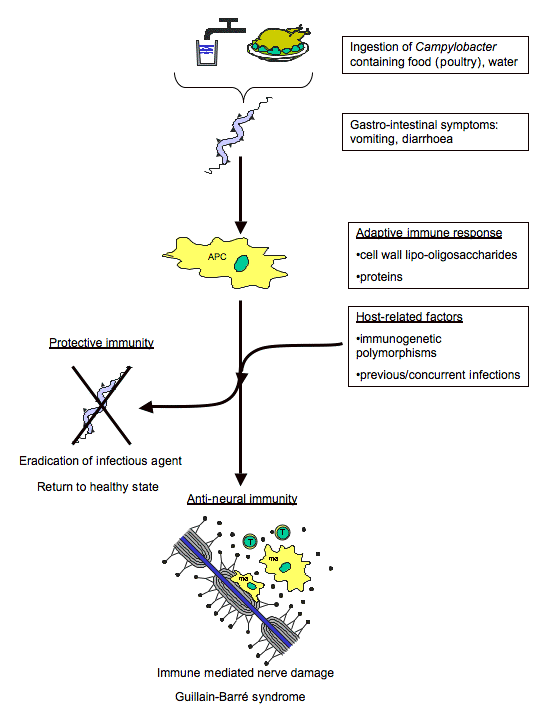
Figure 6. Mechanism of molecular mimicry involved in GBS autoimmune response after C. jejuni infection. (Image from Ang et al., 2004. Reproduced with permission of the author.)
MFS-associated Infection
Infection by C. jejuni if also implicated in the development of MFS by the same mechanism of molecular mimicry. MFS patients present with symptoms similar to other forms of GBS along with problems controling eye movements, or ophthalmoplegia. Autoantibodies have been isolated from these patients that bind to human ganglioside GQ1b as well as the GQ1b epitope present within the LOS of C. jejuni isolated from MFS patients. Also, the dominant C. jejuni serotypes associated with MFS were HS:2 and HS:4. The gene polymorphism associated with the development of anti-GD1b autoantibodies was found to be cstII(Asn51) (Koga et al., 2006). This provides a clear link to the clincal presentation of MFS since the GQ1b ganglioside is found predominantly in human oculomotor nerves (Ang et al., 2004; Kuwabara, 2004).
Effective Treatments
The first effective treatment option for GBS was plasmapheresis, or plasma exchange (PE). This treatment involves removing plasma from the blood and using centrifugal blood separators to remove immune complexes and possible autoanitbodies. The plasma is then reinjected into the patient along with a 5% albumin solution to compensate for lost protein concentration. Many studies have found that this treatment involves high risks and adverse effects for hemodynamically unstable patients (Shahar, 2006). Also, studies found that the most effective number of treatments for moderate and severe cases of GBS was four. A post treatment worsening of symptoms was seen as well in 10% of patients in some studies (Kuwabara, 2004). Risks and conclusions like these began the search for effective and safer treatments.
Another treatment for GBS is IV administration of immunoglobulins (IVIg). The anti-idiotypic antibodies used have been shown to modulate the humoral response in their ability to inhibit autoanibodies and suppress autoantibody production. By inhibiting autoantibodies, the complement-mediated damage can be attenuated. IVIg's also block binding of Fc(gamma) receptors preventing phagocytic damage by macrophages. Studies have shown that an optimal amount of IVIg is 400mg/kg over six days. In comparison studies, PE and IVIg were proven to be similar in overall effectiveness, and no increase in effectiveness was seen by combining therapies. However, it was noted that IVIg was considered safer by its reduced risks and complications (Harel and Shoenfeld, 2005; Shahar, 2006).
References
Ang CW, Jacobs BC, Laman JD. (2004) The Guillain-Barre Syndrome: a true case of molecular mimicry. Trends in Immunology. 25(2):61-66.
Harel M and Yehuda Sheonfeld. (2005) Intravenous Immunoglobulin and Guillain-Barre Syndrome. Clinical Reviews in Allergy & Immunology. 29:281-287.
Ho TW, McKhann GM, Griffin JW. (1998) Human Autoimmune Neuropathies. Annu. Rev. Neurosci. 21:187-226.
Hughes RAC, Allen D, Makowska A, Gregson NA. (2006) Pathogenesis of chronic inflammatory demyelinating polyradiculoneuropathy. Journal of the Peripheral Nervous System. 11:30-46.
Janeway, CA, Travers P, Walport M, Shlomchik MJ. (2005) Immunobiology: The immune system in health and disease, 6th edition. New York: Garland Publishing.
Koga M, Gilbert M, Takahashi M, Li J, Koike S, Hirata K, Yuki N. (2006) Comprehensive analysis of bacterial risk factors for the development of Guillain-Barre Syndrome after Campylobacter jejuni infection. Journal of Infectious Disease. 193:547-55.
Kuwabara S. (2004) Guillain-Barre Syndrome: epidemiology, pathophysiology, and management. Drugs. 64(6):597-610.
Moran PM, Annuk H, Prendergast MM. (2005) Antibodies induced by ganglioside-mimicking Campylobacter jejuni liooligosaccharides recognise epitopes at the nodes of Ranvier. Journal of Neuroimmunology. 165:179-185.
Neisser A, Schwerer B, Bernheimer H, Moran AP. (2000) Ganglioside-induced antiganglioside antibodies from a neuropathy patient cross-react with lipopolysaccharides of Campylobacter jejuni associated Guillain-Barre syndrome. Journal of Neuroimmunolgy. 102:85-88.
Shahar E. (2006) Current therapeutic options in severe Guillain-Barre Syndrome. Clinical Neuropharmacology. 29(1):45-51.
Yuki N. (2005) Carbohydrate mimicry: a new paradigm of autoimmune diseases. Current Opinion in Immunology. 17:577-582.