This web page was produced as an assignment for an undergraduate course at Davidson College.
Hyper-IgM Syndrome (HIGM)
Hyper-IgM Syndrome (HIGM) currently encompasses a group of six diseases associated with immunodeficiency. Mutations resulting in various forms of HIGM syndrome affect the specificities of the antibodies produced by B cells in response to pathogens. Specifically, isotype switching and somatic hypermutation (SHM), two processes following B cell activation, are aberrant in HIGM patients (reviewed in Etzioni and Ochs 2004).
Background Information
Activation of B cells
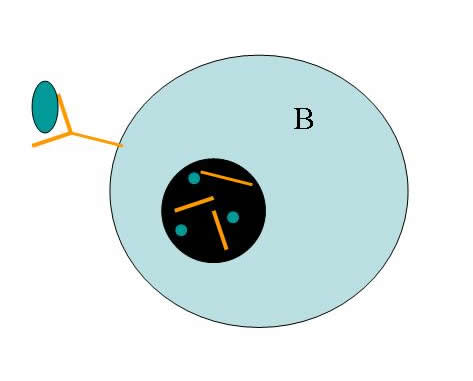
The activation of B cells is necessary for the humoral response to a pathogenic infection. Anchored in the plasma membrane of each mature, unactivated B cell are hundreds of identical B cell receptors (BCRs), which are unique to that B cell (Janeway et al. 2005). Upon recognition of an antigen for which a BCR is specific, the BCR:antigen complex is endocytosed by the B cell (Figure 1).
Figure 1: B cell antigen recognition. When B cell receptors bind the antigen to which they are specific, both the receptor and the antigen are endocytosed. This endosome fuses with a lysosome, causing the B cell receptor and the antigen to be broken down into peptide fragments.
This endosome fuses with a lysosome, which contains proteases to break down the contents of the endosome. Individual peptides resulting from this degradation are loaded into complexes called MHC class II and are presented on the surface of the B cell (Janeway et al. 2005). In the lymph node, a CD4+ T cell with specificity for the presented peptide in the context of MHC class II will bind to the B cell via the MHC:peptide complex (Figure 2). This interaction, along with the interaction of co-stimulatory molecules expressed on lymphocyte surfaces, causes signal transduction cascades within both the B cell and the T cell, triggering activation and proliferation of each lymphocyte (Janeway et al. 2005).
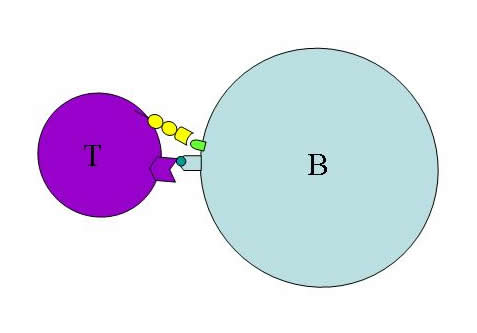
Figure 2: B cell activation. A peptide presented in the context of MHC class II of the B cell is recognized by the T cell receptor. Co-stimulatory molecules also interact. These events cause activation and proliferation of both the B cell and the T cell.
Isotype Switching
Following activation, B cells migrate to the medullary cords of the lymph node and establish a primary focus. The primary focus is a site of B cell proliferation and isotype switching (Janeway et al. 2005). Isotype switching is the recombination of genes encoding the constant regions of antibodies to produce antibodies of class IgG, IgA, IgM, IgE, or IgD (Janeway et al. 2005). Different classes of antibodies are optimized to home to different tissues of the body when secreted. IgG is the most plentiful class of antibody and is present in the blood and the lymph. It is also the only class that can cross the placental barrier to reach a developing fetus. IgA is present in secretions such as saliva, tears, and breast milk. IgM is the least specific immunoglobulin class and is mainly present on the surface of the B cell and in the blood. IgE is present in mucosal epithelium and is responsible for the allergic response as well as responses to parasites. IgD is present mainly on B cell surfaces and is not usually secreted (Janeway et al. 2005). The first istoypes to appear on the surface of the B cell are IgM and IgD, which requires no recombination of the genes encoding the immunoglobulins. Also in the primary focus, B cells begin secreting IgM. After the initial response, the expression of IgM and IgD is arrested, and recombination occurs in the immunoglobulin genes (Figure 3).
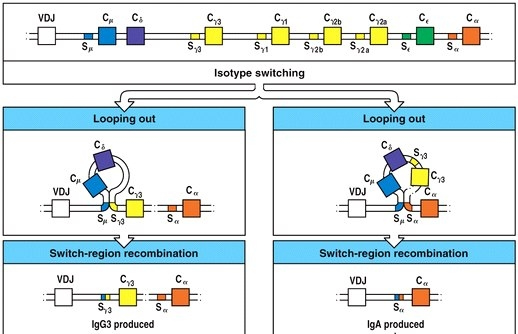
Figure 3: Recombination events in isotype switching. Genes encoding the constant region corresponding to each isotype are arranged in a linear fashion. During isotype switching, a portion of the DNA is looped out as switch regions recombine. After recombination, the constant region directly downstream of the VDJ region will encode the isotype produced on the surface of the B cell and secreted (Image from Janeway et al. 2005, Figure 4.21).
As a result of this recombination, a different isotype is produced which has an ideotype consistant with that of the IgM already secreted. In the end, a mature and activated B cell has immunoglobulins of a single isotype on its surface. Isotype switching enables a more targeted attack against an infection. It does not affect antigen binding, but instead, istotype switching affects where and by what mechanism the humoral response takes place.
Somatic Hypermutation
Another process that strengthens the specificity of the humoral response to pathogenic infection is somatic hypermutation (SHM). After isotype switching, B cells migrate to the primary follicle of the lymph node where they establish a germinal center, which is the primary site for proliferation. While in the germinal center, the variable regions of the genes encoding antibodies undergo SHM (Janeway et al. 2005). During SHM, the variable regions of antibodies have a high mutation rate.
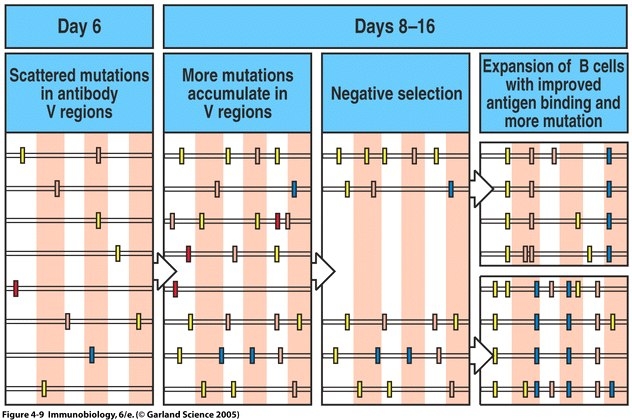
Figure 4: Somatic hypermutation and affinity maturation. In the germinal centers of a lymph node, the variable regions of BCRs are constantly acquiring mutations. Mutations that negatively affect the BCR's affinity for its antigen will be selected against, while mutations in the BCR that positively affect affinity for the antigen will cause the corresponding B cell to proliferate (Image from Janeway et al. 2005, Figure 4.9).
Those alterations in the variable region producing a lower affinity for the antigen are selected against, while mutations producing a higher affinity for the antigen continue to proliferate (Janeway et al. 2005; Figure 4). Therefore, as time elapses within the humoral immune response, the antibodies secreted by B cells become more specific for their antigen.
Wild Type Pathway to Class Switch Recombination and Somatic Hypermutation
The interaction between a B cell and a T cell that are specific for the same antigen triggers a cascade of events which ends with isotype switching and SHM. On the surface of the B cell, the MHC class II complex binds to the T cell receptor. Co-stimulatory signals are provided through the interaction of CD40 on the B cell surface and CD40 Ligand (CD40L) on the T cell surface (Janeway et al. 2005). These two interactions on the surface cause the activation of the NFκB essential modulator (NEMO). NEMO is responsible for phosphorylating IκB, which usually acts as an inhibitor for the transcription factor, NFκB. Phosphorylation of IκB causes its degradation, freeing NFκB to translocate to the nucleus and begin transcription of essential genes for the processes of isotype switching and SHM (reviewed in Etzioni and Ochs 2004). Two of these genes, activation-induced cytidine deaminase (AICDA) and uracil N-glycosylase (UNG), are known to perform essential functions leading to isotype switching and SHM. During transcription, AICDA, the gene encoding AID, is responsible for deaminating cytosines within the single stranded DNA encoding immunoglobulins (Revy et al. 2000). Following this, UNG recognizes the sites of deamination and cleaves this base. These cleavages, when occurring in close proximity on the template and non-template strand, allow for isotype switching and SHM (Kavli et al. 2005). The exact steps following base excision by UNG are yet to be delineated. In each form of hyper-IgM syndrome, a protein in this cascade is not functional, disrupting the pathway to isotype switching and SHM (reviewed in Etzioni and Ochs 2004; Figure 5).
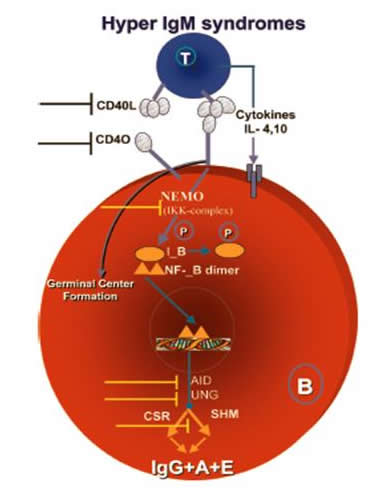
Figure 5: Signal transduction pathway to istoype switching and SHM and disruptions in hyper-IgM syndromes. When a B cell and a T cell recognize the same antigen, a signal transduction pathway is initiated within the B cell, which leads to isotype switching (synonymous with class switch recombination, or CSR) and somatic hypermutation (SHM)(Image from Etzioni and Ochs 2004, permission pending).
Hyper-IgM Syndrome
Hyper-IgM Syndrome (HIGM) is a relatively rare group of diseases resulting from mutations in the pathway from B-cell activation to isotype switching and SHM. Patients with HIGM are usually diagnosed within the first two years of life and experience severe immunosuppression, owing to decreased levels of serum IgG, IgA, and IgE (reviewed in Etzioni and Ochs 2004). Treatment options currently include intravenous supplement of immunoglobulins for which the patient is deficient as well as bone marrow transplants. Depending on the mutation causing this syndrome, different modes of inheritance are seen. The severity of phenotypes within each form of HIGM varies widely due to variability in the mutations causing the disease (Serra et al. 2001 ; Lopez-Granados et al. 2003).
Hyper-IgM Type 1
HIGM1 is an X-linked recessive disease, with the majority of cases occurring in males. Female carriers do not experience immunosuppression and undergo normal isotype switching and SHM (reviewed in Etzioni and Ochs 2004). Patients with HIGM1 are at high risk for recurrent infections by opportunistic pathogens, liver disease, and neutropenia (Levy et al. 1997). The HIGM1 phenotype results from mutations in CD40L, a co-stimulatory molecule expressed on the surfaces of T cells (Brezinschek et al. 2000; Andre et al. 2002; Figure 5). Without this co-stimulation, B cells do not receive the needed signal to undergo isotype switching and SHM. Germinal centers also fail to develop in lymph nodes of HIGM1 patients (Subauste et al. 2002; Jain et al. 2002). Therefore, the only antibody secreted by the B cell is IgM, the least specific class of antibody.
Hyper-IgM Type 2
HIGM2 is an autosomal recessive disease characterized by mutations in activation-induced cytidine deaminase, AICDA (Figure 5). The gene product of AICDA is AID. Unlike HIGM1, HIGM2 patients are not frequently infected by opportunistic pathogens (Revy et al. 2000), presumably because T-cell-mediated immunity is still functioning correctly. However, HIGM2 patients generally develop lymphoid hyperplasia (Revy et al. 2000). Because germinal centers are able to develop in the lymph node, but no SHM occurs, there is no selection process of the proliferating B cells according to antigenic affinity. As a result, giant germinal centers are seen in the lymph nodes of HIGM2 patients (Revy et al. 2000).
Hyper-IgM Type 3
The phenotype in HIGM3 is similar to that of HIGM1 because mutations occur in CD40, a co-stimulatory molecule on the surface of B cells that interacts with CD40L on T-cell surfaces (Fontana et al. 2003; Figure 5). Like HIGM1 patients, HIGM3 patients fail to undergo isotype switching and SHM, do not form germinal centers, and are subject to frequent infections by opportunistic pathogens (Fontana et al. 2003). The mode of inheritance for HIGM3 is autosomal recessive.
Hyper-IgM Type 4
HIGM4 is the least-characterized of the HIGM types. Unlike other forms of HIGM, HIGM4 patients show a milder immunosuppressance and have residual levels of IgG (McLean et al. 2004; Darlow et al. 2004). Many HIGM4 patients were originally diagnosed with HIGM2 until it was found that AID functioned normally in these patients, indicating that the mutation in HIGM4 patients occurs in a gene active downstream of AID in the B-cell activation pathway (Imai et al. 2003). HIGM4 patients often experience lymphoid hyperplasia, but do not manifest enlarged germinal centers (reviewed in Etzioni and Ochs 2004). SHM has also been observed in HIGM4 patients, suggesting that the mutation in HIGM4 may be isotype switching-specific (Manis and Alt, 2003). More specifically, mutations in HIGM4 patients may interrupt survival signals given to B cells by the antigen following isotype switching, causing B cells of classes other than IgM to undergo apoptosis (Imai et al. 2003). Alternatively, the mutation may prevent the ligation of DNA strands following isotype switching (Imai et al. 2003). The mode of inheritance for HIGM4 is proposed to be autosomal recessive, although many cases have arisen through de novo mutations (Imai et al. 2003).
Hyper-IgM Type 5
HIGM5 is caused by a mutation in the catalytic region of the uracil DNA glycosylase (UNG) gene (Figure 5). UNG is responsible for the cleavage of cytosines that have been deaminated by AID in single-stranded DNA. A mutation in the UNG gene prevents the base cleavage necessary for both isotype switching and SHM (Kavli et al. 2005). Because of this, patients with HIGM5 have increased levels of deaminated cytidine within the genome. This autosomal recessive disease is clinically similar to HIGM2, with incidences of lymphoid hyperplasia and recurrent bacterial infections (Kavli et al. 2005).
Hyper-IgM Type 6
A second X-linked recessive form of HIGM, HIGM6, was first discovered as a form of the syndrome, EDA (ectodysplasin-A). It was found that some forms of EDA were associated with immunodeficiency and were named EDA-ID. EDA-ID is now synonymous with HIGM6 (reviewed in Etzioni and Ochs 2004). The mutation causing HIGM6 is in the NEMO (nuclear factor κB essential modulator) complex (Figure 5), which, when mutated, is unable to phosphorylate IκB (reviewed in Etzioni and Ochs 2004). Therefore, transcription is never initiated by NFκB, and isotype switching and SHM cannot occur.
Summary
Hyper-IgM Syndrome is a great example of the necessity of many proteins working together in keeping the body healthy. A mutation in any of the major players of the pathway to isotype switching or SHM can result in immunosupression. However, treatments for this group of diseases are available. HIGM is also demonstrative of the importance of isotype switching and somatic hypermutation to the humoral immune response. As more is found out about the mechanism of action of each of the proteins mutated in the forms of HIGM, more information can also be elucidated concerning the humoral immune response.
Works Cited:
Andre, P., Srinivasa Prasad, K.S., Denis, C., He, M., Papalia, J.M., Hynes, R.O., Phillips, D.R., and Wagner, D.D. 2002. CD40L stabilizes arterial thrombi by a beta-3 integrin-dependent mechanism. Nature Medicine 8: 247-252.
Brezinschek, H.P., Dorner, T., Monson, N.L., Brezinschek, R.I., and Lipsky, P.E. 1999. The influence of CD40-CD154 interactions on the expressed human VH repertoire: analysis of VH genes expressed by individual B cells of a patient with X-linked hyper-IgM syndrome. International Immunology 12: 767-775.
Costa-Carvalho, B.T., Viana, M.A., Brunialti, M.K.C., Kallas, E.G., and Somallo, R. 2004. An imbalance of naïve and memory/effector subsets and altered expression of CD38 on T lymphocytes in two girls with hyper-IgM syndrome. Clinical and Experimental Immunology 136: 291-296.
Darlow, John M., Farrell, Alex M., and Stott, David I. 2004. Non-functional immunoglobulin G transcripts in a case of hyper-immunoglobulin M syndrome similar to type 4. Immunology 111: 212-222.
Etzoni, Amos and Ochs, Hans D. 2004. The hyper IgM syndrome—An evolving story. Pediatric Research 56: 519-525.
Fontana, S., Moratto, D., Mangal, S., De Francesco, M., Vermi, W., Ferrari, S., Facchetti, F., Kutukculer, N., Fiorini, C., Duse, M., Das, P.K., Notarangelo, L.D., Plebani, A., and Badolato, R. 2003. Functional defects of dendritic cells in patients with CD40 deficiency. Blood 102: 4099-4106.
Granados, E.L., Cambronero, R., Ferreira, A., Fontan, G., and Garcia-Rodriguez, M.C. 2003. Three novel mutations reflect the variety of defects causing phenotypically diverse X-linked hyper IgM syndrome. Clinical and Experimental Immunology 133: 123-131.
Imai, K., Catalan, N., Plebani, A., Marodi, L., Sanal, O., Kumaki, S., Nagendran, V., Wood, P., Glastre, C., Sarrot-Reynauld, F., Hermine, O., Forveille, M., Revy, P., Fischer, Alain., and Durandy, A. 2003. Hyper-IgM syndrome type 4 with a B lymphocyte-intrinsic selective deficiency in Ig class-switch recombination. Journal of Clinical Investigation 112: 136-142.
Jain, A., Prescott Atkinson, T., Lipsky, P.E., Slater, J.E., Nelson, D.L., and Strober, W. 1999. Defects of T-cell effector function and post-thymic maturation in X-linked hyper-IgM syndrome. The Journal of Clinical Investigation 103: 1151-1158.
Janeway, C.A., Travers, P., Walport, M., and Shlomchik, M.J. 2005. Immunobiology, Sixth Edition. Garland Science, 821.
Kavli, B., Anderson, S., Otterlei, M., Liabakk, N.B., Imai, K., Fischer, A., Durandy, A.,
Krokan, H.E., and Slupphaug, G. 2005. B cells from hyper-IgM patients carrying UNG mutations lack ability to remove uracil from ssDNA and have elevated genomic uracil. JEM 201(12): 2011-2021.
Levy, J., Espanol-Boren, T., Thomas, C., Fischer, A., Tovo, P., Bordigoni, P., Resnick, I. , Fasth, A., Baer, M., Gomez, L., Sanders, E. A. M., Tabone, M.-D., and 13 others. 1997. Clinical spectrum of X-linked hyper-IgM syndrome. J. Pediat. 131: 47-54.
Manis, John P., and Alt, Frederick W. 2003. Novel antibody switching defects in human patients. The Journal of Clinical Investigation 112: 19-22.
McLean, G.R., Miller, K.K., Schrader, J.W., and Junker, A.K. 2004. Biased immunoglobulin G (IgG) subclass production in a case of hyper-IgM syndrome. Clinical and Diagnostic Laboratory Immunology 11: 1192-1193.
Revy, P., Muto, T., Levy, Y., Geissmann, F., Plebani, A., Sanal, O., Catalan, N., Forveille, M., Dufourcq-Lagelouse, R., Gennery, A., Tezcan, I. , Ersoy, F., and 9 others. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell 102: 565- 575.
Serra, H.M., Baena-Cagnani, C.E., Mariani, A.L., Martin, A., Garip, E.A, and Pesoa, S.A. 2001. A new CD154 mutation. Allergy 56: 1230-1238.
Subauste, C.S., Wessendarp, M.W., Sorenson, R.U., and Leiva, L.E. 1999. CD40-CD40 ligand interaction is central to cell-mediated immunity against Toxoplasma gondii: patients with hyper-IgM syndrome have a defective type 1 immune response that can be restored by soluble CD40 ligand trimer. The Journal of Immunology 162: 6690-6670.
Walace, A.J., Wu, C.L., and Elles, R.G. 1999. Meta-PCR: a novel method for creating chimeric DNA molecules and increasing the productivity of mutation scanning techniques. Genet. Test. 3: 173-183.
If you have any questions or concerns about this web page please contact Sarah.
Return to Davidson College Biology Home Page.
Return to Immunology Home Page.
Return to Sarah's Immunology Home Page.