This page was created as an assignment for an undergraduate class at Davidson College
Overview of the Complement System
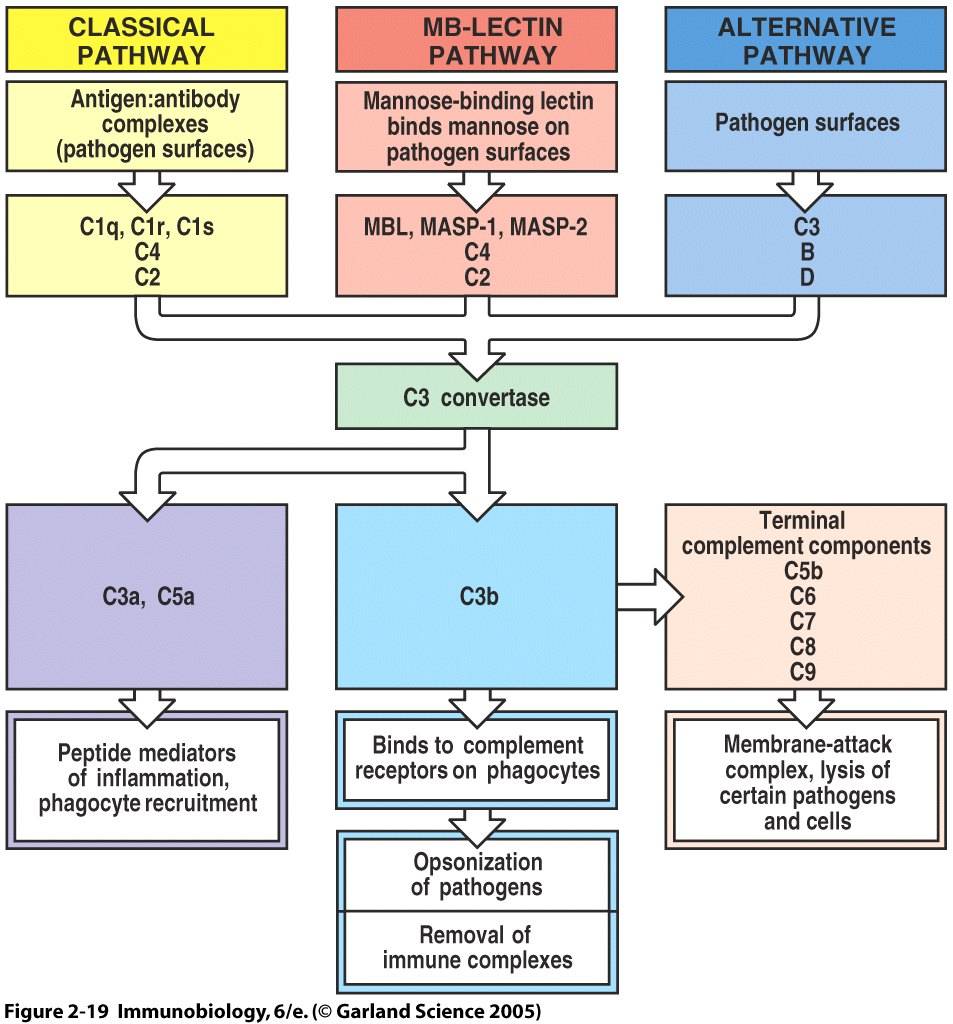
The complement system is a component of innate immunity that consists of multiple plasma proteins which act to fight infection by opsonizing pathogens, inducing inflammatory responses, enhancing antibody responses, and attacking some pathogens directly. Activation of the complement cascade relies on cleavage of a zymogen to yield an active enzyme that in turns cleaves and activates the next zymogen in the cascade. Through this series of cleavage and enzyme activation, the immune system is able to produce a wide-reaching response to few stimulation events. Three pathways which can activate the complement system are ounlined below and depicted in Figure 1. The three pathways converge at the generation of a C3 convertase. Cleavage of C3 is the most important step in the activation pathway. Large numbers of opsonizing proteins, primarily C3b, are generated and coat pathogen surfaces to facilitate their uptake and destruction by phagocytic cells. Small peptide mediators of inflammation are released, leading to the increased migration of phagocytes to the site of infection. In addition, terminal complement components are able to form membrane-attack complexes and destroy certain pathogens by disrupting their membrane integrity (Janeway et al., 2005). This website focuses on the activation, effector function, and deficiency of C3. C3 is the most abundant protein in the complement system, with a serum concentration of approximately 1.2 mg/mL (Sahu and Lambris, 2001).
Figure 1. Overview of Complement Activation and Function. All three pathways that activate the complement cascade converge at the formation of a C3 convertase. This complex cleaves C3 into components C3a and C3b, ultimately leading to pathogen opsonization, release of inflammatory mediators, and formation of terminal complement components.
In the classical pathway, complement protein C1q binds to antibody:antigen complexes on pathogen surfaces. Conformational changes in the C1 complex transforms C1s into its active protease form, which cleaves C4 and C2 proteins. The C4b and C2b combine to generate a C4b2b complex. C4b2b is an active C3 convertase that cleaves C3 into C3a and C3b. This process is summarized in Figure 2 (Janeway et al., 2005). C4b2b is only functional in the presence of magnesium and decays over time, factors which we will see are important to complement control (Merck, 2006).
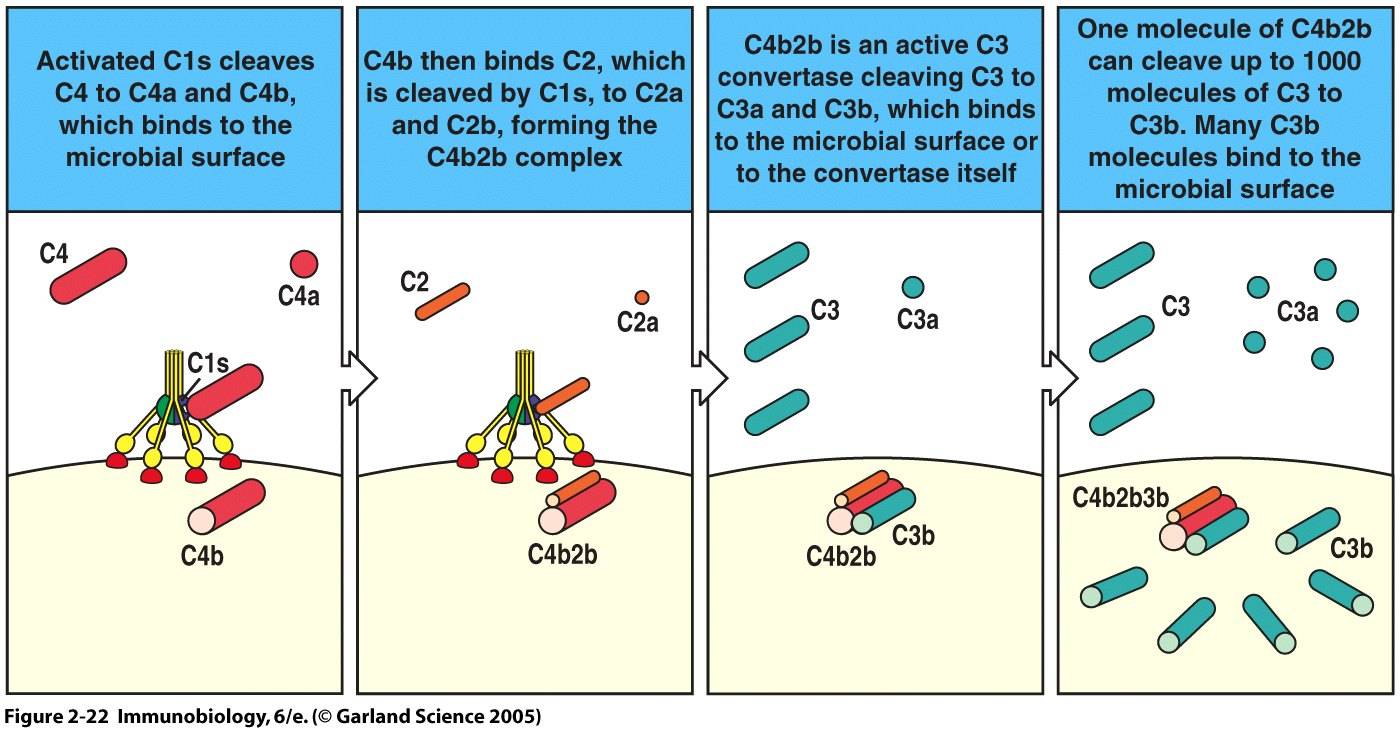
Figure 2. Classical Pathway of Complement Activation. Binding of C1 to a pathogen surface initiates an enzyme cascade which culminates in the formation of a C3 convertase and generation of effector molecules C3a and C3b.
The mannose-binding lectin pathway begins when the serum protein lectin binds to mannose residues expressed in a pattern unique to pathogen surfaces. Zymogen proteases MASP-1 and MASP-2 are activated and lead to the formation of a C3 convertase from C4 and C2.
The alternative pathway is unique in that it does not require an antigen-binding protein. Some C3 undergoes spontaneous hydrolysis to yield C3(H2O). It is estimated that at any given instant, about 0.5% of total C3 exists in this hydrolyzed form (Sahu and Lambris, 2001). The plasma protein factor B binds to the C3(H2O) complex and is subsequently cleaved by factor D to form a C3(H2O)Bb complex. This is a C3 convertase, which cleaves large numbers of additional C3 into its components. C3bBb also requires magnesium and decays over time (Merck, 2006). The alternative pathway is viewed as an amplification loop because C3b deposited on pathogen surfaces by the classical or mannose-bindign lectin pathways can combine with factors B and D to form C3bBb. One C3 convertase complex can cleave numerous C3 molecules and amplify the response (Janeway et al., 2005). All of these pathways converge at the formation of a C3 convertase, therefore we see that the cleavage of C3 into C3a and C3b is the pivotal point in complement response.
Figure 3. Wireframe Structure of C3. C3 is a large molecular weight protein with several cleavage sites that separate it into multiple components, including C3a, C3b, C3dg, C3c, and so forth. It contains many functional domains that allow it to interact with more than 25 proteins (Janssen et al., 2005)
The C3 locus has been traced to a 41 kb gene on chromosome 19 by a series of experiments using monoclonal antibody with specificity for human C3 and following continued expression of this gene through different strains of human-mouse somatic cell hybrids. This locus was further confirmed by using a cDNA probe for the gene in DNA hybridization experiments (Whitehead et al., 1982). It is noteworthy that this complement protein segregates independently of the major histocompatibility complex, located on chromosome 6.
The complement proteins are capable of inducing strong responses on cells they bind to, therefore the target and lifetime of these proteins muct be carefully regulated. Many cleavage products of these proteins, such as C3b, are quickly inactivated unless they bind to pathogen surfaces. Other products are stable only when complexed with other complement proteins. C3bBb, for example, must be stabilized by the regulatory protein properdin (factor P) on pathogen surfaces to continue its cleavage of C3 molecules (Janeway et al., 2005).
Complement proteins do not have a direct mechanism of distinguishing between host and foreign cells. Inadvertant activation of complement on host cells can lead to damage of necessary cells. Due to this threat, the body has evolved ways to protect it's cells. C3b has binding sites for multiple complement proteins that either amplify or inactivate C3b. Host cells are protected against complement activity by regulatory proteins which quickly destroy complement components deposited on their surface. Factor I circulates and cleaves any C3b that has bound membrane cofactor proteins on host cells into its inactive forms iC3b and C3dg in the presence of factor H, CR1, and membrane co-factor protein. Pathogen surfaces, on the other hand, lack these regulatory proteins, allowing complement proteins are stabilized by molecules like properdin. In addition, C3 convertase activity is amplified by factor B in the presence of factor D (Sahu and Lambris, 2001). Other proteins, such as the decay-accelerating factor (DAF), regulate the classical and alternative pathways by breaking down C3 convertases (Janeway et al., 2005).
While the body has many effective ways of regulating complement activation, a current area of research is the application of complement inhibitors in drugs that could stop complement-mediated damage to host cells. Compstatin is a protein that does not exist naturally in the body but binds to naive C3 and blocks its cleavage by C3 convertase. Experiments have suggested that using compstatin to inhibit complement can increase tolerance to transplanted organs (Sahu and Lambris, 2001).
A single active C3 convertase is capable of depositing up to one thousand C3b molecules on a pathogen surface. Cleavage of C3 into C3b exposes a highly reactive thioester bond which reacts with hydroxyl groups on nearby surface molecules. Since pathogen surfaces lack deactivating control factors, opsinizing proteins are stabilized.
C3b binds to many receptors on phagocytic cells and stimulates these cells to ingest the tagged pathogen. The first of these complement receptors, CR1, activates macrophages to undergo phagocytosis in the presence of other immune mediators. CR1 is also found on the surface of red blood cells. In this context, the red blood cell is able to bind C3b attached to antigen: antibody complexes and carry them to the spleen or liver, where phagocytic cells remove and eliminate the complexes. A defect in this eliminatory function can lead to systemic lupus erythematosus. Integrin receptors CR3 and CR4 differ in that they bind to the inactive form iC3b (cleaved by factor I) still remaining on pathogen surfaces. Binding through these receptors are sufficient to stimulate phagocytosis without co-stimulation by other proteins (Janeway et al., 2005).
CR2 is found on B cells, follicular dendritic cells, and some T cells. The receptor serves as part of the co-receptor complex on B cells by binding C3dg, an alternate cleavage product of C3b. B cells that bind a specific antigen to their receptor receive a much stronger activation signal if they also bind C3dg via CR2. This mechanism allows the complement system to strengthen the antibody response of the acquired immune system (Janeway et al., 2001). C3 knockout mice display fewer and smaller germinal centers and impaired antibody responses (Sahu and Lambris, 2001).
We have seen that the cleavage of C3 is the central event in the complement response, therefore it must be able to activate later components of the complement system (Fig. 1). C3b fragments bind to their C3 convertases (C4b2b in the classical and lectin pathways and C3bBb in the alternative pathway) to form C5 convertases. C5 is then cleaved by C2b or CBb to yield large amounts of C5a and C5b. C5a is the most potent peptide mediator of inflammation. In addition, it serves as a chemoattractant to recruit more phagocytes to the site of infection.
C5b initiates the construction of a membrane pore from complement proteins C6, C7, C8, and C9. Each protein adds to teh complex in turn and multiple C9 molecules insert themselves into the lipid-bilayer membrane of certain pathogens. The resulting membrane-attack complex is devastating to the cell because it disrupts membrane integrity. Loss of chemical gradients and selective permeability ultimately lead to lysis and destruction of the cell (Janeway et al., 2005).
Receptors for C3a and C5a are present on the surface of enothelial cells, mast cells, and phagocytes. Binding of the peptide ligands to their seven-span transmembrane receptors leads to the activation of G-proteins and signaling through secondary messengers in the interior of the cell. A local inflammatory response ensues. Blood flow and vascular permeability are increased and adhesion molecules are expressed to facilitate the migration of neutrophils and monocytes to the infected tissues. C3a and C5a also induce mast cells to histamine and TNF-alpha, which contribute to the proliferation of the inflammatory response (Janeway et al.,2005).
Deployment of the complement system is one of the body's main defenses against viral infection. Activation of the complement cascade can lead to augmentation of the antibody response, phagocytosis of opsonized cells, lysis of infected cells through the membrane-attack complex, and inflammation. Complement proteins support both the innate and adaptive immune responses, and inhibition of any part of the complement casacade can make an individual much more susceptible to infection by viruses or certain bacteria (Sahu and Lambris, 2001).
Deficiencies or mutations in the third complement protein devastate the entire complement system. Mutations in C3 are rare and resulting conditions are inherited in an autosomal recessive fassion. While complement deficiencies are rare, patients with deficiencies in C3 present with frequent infections at a young age and suffer a high mortality rate. C3 deficiencies inhibit the opsinozation of pathogenic surfaces, limit the chemotaxis of leukocytes to sites of infection, and allow for the survival of pathogens that would normally be destroyed by the membrane-attack complex (e.g. meningitis). As a result, 79% of patients with a C3 deficiency develop collagen vascular disease. There is no specific treatment for patients with complement deficiencies other than treating opportunistic infections as they arise (Chaganti, 2006).
Patients with C3 deficiencies are predisposed to autoimmune diseases such as systemic lupus erythematosus. Circulating immune complexes are not cleared through interactions between C3b and CR1 on red blood cells. As immune complexes accumulate and increase in concentration they lead to tissue damage (Chaganti, 2006). Many parts of the body can be affected, including blood vessels, heart, lungs, brain, joints, and skin. Common symptoms include fatigue, swollen or painful joints, fever, rahes, and kidney complications (NIAMS, 2003).
Due toC3's central role in the host's response to infection, many microorganisms have become pathogenic by developing mechanisms to evade or inhibit the action of C3. This is accomplished by using the host's protein synthesis apparatus to produce homologues of human complement control proteins. The vaccinia virus, which leads to progressive necrosis (tissue death), encodes a vaccinia virus complement control protein (VCP) that inactivates complement by enhancing cleavage of C3b and C4b by factor I. Vaccinia is a pox virus and was used in vaccinations to confer immunity against smallpox (Lachman, 2002). Herpes simplex viruses types 1 and 2 produce glycoprotein C molecules that bind and inhibit C3b on pathogen surfaces and accelerates the decay of C3 convertase C3bBb by blocking properdin binding (Sahu and Lambris, 2001).
Changanti, Krishna. "Complement Deficiencies." eMedicine, 2006. http://www.emedicine.com/med/topic419.htm
Janeway, C.A., Travers, P., Walport, M., Shlomcik, M. Immunobiology: The Immune System In Health and Disease. 6th ed. New York: Garland Science, 2005.
Lachman, P.J. "Microbial Subversion of teh Immune Response." Proceedings of the National Academy of Sciences of the United States of America. Vol. 99, No.13. Jun. 2002: 8461-8462.
Whitehead, Alexander et al. "Assignment of the Structural Gene for the Third Component of Human Complement to Chromosome 19." Proceedings of the National Academy of Sciences of the United States of America. Vol. 79, No.16. Aug. 1982: 5021-5025.
The Merck Manual of Diagnosis and Therapy. Section 12, Ch. 146: Biology of the Immune System. http://www.merck.com/mrkshared/mmanual/section12/chapter146/146d.jsp
Sahu, Arvind and John D. Lambris. "Structure and biology of complement protein C3, a connecting link between innate and acquired immunity." Immunological Reviews. Vol. 80. 2001: 35-48.
National Institutes of Health: National Institute of Arthritis and Musculoskeletal and Skin Diseases. NIH Publication No. 03-4178, 2003. http://www.niams.nih.gov/hi/topics/lupus/slehandout/index.htm
Janssen, B.J.C., Huizinga, E.G., Raaijmakers, H.C.A., Roos, A., Daha, M.R., Nilsson-Ekdahl, K., Nilsson, B., Gros, P. "Structures of complement component C3 provide insights into the function and evolution of immunity." Nature Vol. 437, 2005: pp.505-511. http://www.rcsb.org/pdb/explore.do?structureId=2A73
![]()
![]()