Figure 1. Jmol representation of C5a molecular structure. C5a acts as one of the body's most potent mediators of inflammation with its receptor CD88. (PDB image, Zhang et al., 1997)
Introduction
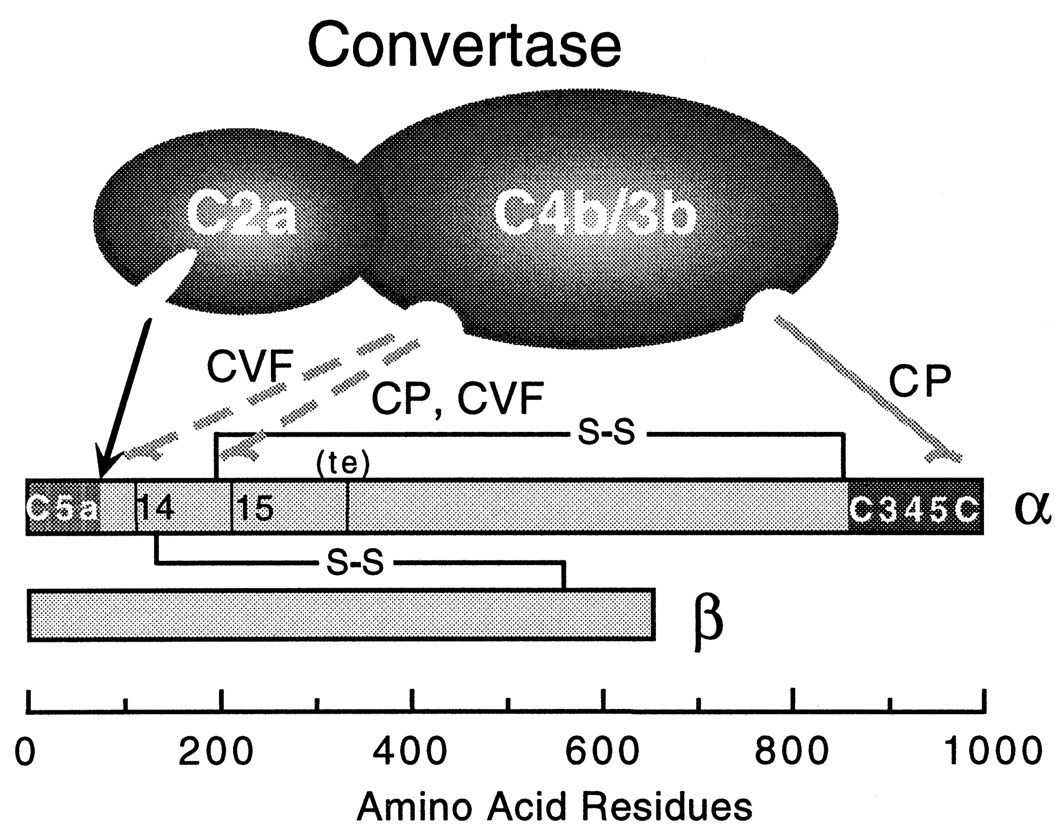
Good health relies on a strong immune system to fight threatening pathogens from infecting host cells. The complement system consists of a series of plasma protein interactions which mediate direct attack on pathogens (opsonization) and induce inflammation. Most complement proteins are zymogens which are proteases that become activated when cleaved. Each of the three pathways of the complement system results in the formation of C3 convertase on the pathogen surface. The C3 convertase complex cleaves C3 into C3a and C3b. C3b is a membrane associated protein which binds covalently to other complement proteins already on the pathogen surface. The complex of complement proteins on the cell surface after C3b binds forms the C5 convertase. In the classical pathway C5 convertase is composed of C4b2a3b and in the alternative pathway is C3bBbC3b. Both C5 convertases perform the same function, which is to cleave C5 into its two active components C5a and C5b. The C5 molecule has a molecular weight of about 190 kDa and consists of two polypeptide chains (α, 115 kDa and β, 75 kDa) which are connected by disulfide bonds. The C5 convertase cleaves C5 with one proteolytic cleavage at an arginine residue 75 residues downstream from the C 5 α-chain N terminus (Sandoval et at., 2000). The result of this cleavage is the release of a C5a fragment, a potent inflammatory molecule, and activation of C5b which initiates the membrane attack complex (MAC). C5 plays a significant role in vital immunolgical pathways as well as contributing to several human diseases.
C5b
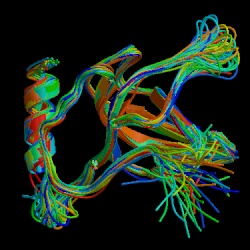
The C5b protein consists of an α (104 kDa) and a β chain (75 kDa). After cleavage C5b interacts nonenzymatically with other complement proteins to form the MAC. The C5b fragment first exposes a binding site having high affinity for C6 and C7 with an associated half life of 2.3 minutes (Dalmasso, 1998). The binding domain of C5b for other complement proteins such as C6 and C7 has been recently characterized and is homologous to a complement binding domain on C3 and C4. This active binding site is C5-C345C (Bramham et al., 2004) and is located about 800 amino acid residues away from the C5 cleavage site toward the N terminus of the α chain (see figure 3). C5-C345C also interacts with C5 convertase although it is not the cleavage site. This site is thought to be the most active site for binding during MAC initiation because binding of C7 is essential for nonreversible MAC formation. The structure of the C5-C345C domain is illustrated in Figure 2.
Figure 2. NMR structural depiction of C5-C345C. The most interactive binding site of C5b is at the C5-C345C domain. C5 convertase cleaves C5 about 800 amino acids away from C345C. Other complement proteins such as C6 and C7 interact with C5 at the active site C345C in forming the MAC. (PDB image, Bramham et al., 2005)
Figure 3. This diagram of C5 convertase shows where C5 is cleaved (dotted arrows) to become activated and where the functional C5-C345C domain is relative to the cleavage site. C5 consists of two chains, but most of the funcional activity for C5 converase and other complement binding occurs on the α-chain. (image from Sandoval et al., 2000, permission pending)
Membrane Attack Complex
The main function of the MAC is to kill pathogens by disrupting the proton gradient of the cell membrane through lysis. By killing extracellular pathogens with the MAC, endocytosis and therefore host cell infection is prevented. C5b begins the assembly of complement components by binding C6 and C7 to its C5-C345C domain, creating a C5b-7 complex which remains loosely associated with C3b (Dalmasso, 1998). This binding leads to a conformational change of the C7 molecule which exposes a hydrophobic binding site and allows the complex to insert itself into the lipid bilayer of the pathogen. One immediate check for the MAC formation is C3b which directs C5b-7 to the pathogen cell membrane or causes the complex to detach and inactivate. The hydrophobic anchor of C7 in the membrane has a high affinity for phospholipids which ensures strong binding while other complement constituents of the MAC are brought together. The next step in MAC formation involves C5b-C7 interaction with the beta chain of C8 to form C5b-8. This newly formed complex has a greater phospholipid affinity than C5b-7 which has already bound to the pathogen surface. The C8 α chain penetrates the lipid bilayer, forming a transient ion channel pore. This porous opening causes leakage of intracellular potassium which is followed by leakage of amino acids and ribonucleotides (Stevens et al., 2003). The final addition to the MAC is C9 which polymerizes in a large pore formation after binding to C8 α. C9 forms in circular formation on the pathogen surface causing cellular contents to burst out of the cell. Figure 4 illustrates MAC formation.
Because C5b is the principle initiator of the membrane attack complex, it is plays an important role in MAC regulation. If a complement cascade is not stopped before C5 convertase formation, regulation is provided by the lability of the C5b molecule before it can form C5b-7. The two minute window ensures that a MAC will only be formed if other complement components are in place to stablize C5b. Also, CD59 is a membrane-associated inhibitor which interacts with membrane-bound C5b-8 to prevent C9 polymerization (Dalmasso, 1998).
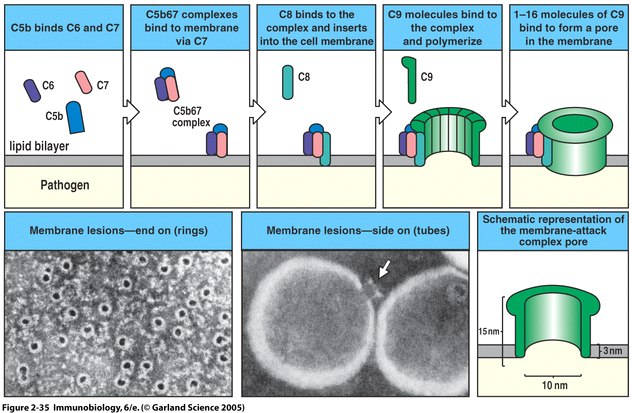
Figure 4. The membrane attack complex is initiated by clustering of C5b-7 which then is inserted into the membrane. Eventually a 10 nm diameter lytic pore is formed which leaks the intracellular contents. C5b is unstable unless bound to C6 so the construction of the MAC is highly dependent upon C5b-7 formation. (Image from Janeway et al., 2005)
C5a
The smaller fragment resulting from C5 cleavage is C5a (77-74 amino acids in length) which is a potent inflammatory molecule. C5a is molecularly characterized by having three disulfide bridges at its core with a disordered C-terminus (see figure 5). As observed in other four-helix protein coiled-coil arrangements, the first 63 residues of C5a in solution form an antiparallel bundle of four helices (Dahinden, 1998). The N-terminus helix is amphiphilic showing interdigitation of hydrophobic residues in helices II and IV (Dahinden, 1998). A molecular arrangement similar to C5a is observed for C3a as well. C5a is highly conserved among most organisms, as are most cytokines, but human C5a has a few molecular distinctions. For example, there is an N-terminal odd cysteine at position 27 in a loop between helices II and III which faces toward the solvent. This distinction may allow for covalent linkage to other proteins with an -SH group (Dahinden, 1998). Also the C-terminus is heavily glycosylated which may explain the chemoattractant properties of human C5a.
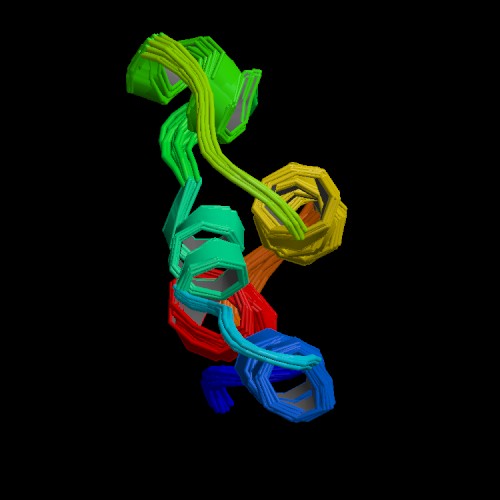
Figure 5. The NMR depiction of the C5a fragment derived from cleavage of C5 is 74 residues in length. Three disulfide bonds link the core of C5a. Arginine residues near the C-terminus determine the agonist capacity of C5a. (PDB image, Zhang et al., 1997)
C5a-R
To understand the powerful biological effects of C5a on the immune system, it is important to consider the interactions between C5a and its receptor C5a-R (CD88). The seven-transmembrane receptor C5a-R (47 kDa) is a G protein-linked receptor in the chemokine receptor family. A two-site binding model of ligand and receptor has been proposed based on homology of C5a-R and the fMLP receptor (Dahinden, 1998). The two-site binding model suggests:
1.) Receptor N-terminal recognition of C5a N-terminal and disulfide-linked core causes conformational change in C5a.
2.) Interaction between C-terminal arginine of C5a with Arg206 on the fifth transmembrane helix of the receptor (Dahinden, 1998).
This model explains why C5a is unable to induce cellular activation when there is no C-terminal arginine (C5adesarg), yet it can still function as a potent chemoattractant, indicating more than one binding interaction with its receptor. Serum carboxypeptidase N is thought to regulate C5a by removing the arginine which converts C5a into C5adesarg. C5a-R is expressed on mast cells, dendritic cells, basophils, monocytes, eosinophils, macrophages, bronchial and alveolar epithelial cells, hepatocytes, astrocytes, neutrophils and vascular endothelial cells.
Anaphylatoxin and Chemotaxin
On binding to its receptor on basophis and mast cells C5a induces histamine release which increases vascular permeability and smooth muscle contraction. In neutrophils C5a induces release of hydrolytic enymes, oxygen radicals, and prostaglandins, calcium transients, shape change, cell polarization, upregulation of receptors, integrin activation, hyperadhesive state, and enhancement of phagocytic and cytotoxic functions (Dahinden, 1998 and Sevens et al., 2003). In granulocytes C5a causes the release of lysosomal enzymes (Cann, 2004). C5a and other small peptides with such potent biological activity are called anaphylatoxins. Of the known anaphylatoxins C5a is the most potent, showing at least 100 times more biological activity than C3a and C4a (Stevens et al., 2003).
C5a acts as an agonist peptide on polymorphonuclear leukocytes which directs them along a chemokine gradient (chemokinesis) to the site of pathogenic infection. Through chemotaxis the increased vascular permeability caused by C5a allows easy migration of neutrophils from blood vessels to tissue. C5a causes monocytes to undergo oxidative burst which releases neutrophil chemotactic factor, platelet activating factor, and interleukin-1 which produces fever and acute phase reactants (Dahinden, 1998 and Stevens et al., 2003). Both are important properties of the inflammatory response. As mentioned these potent biological consequences of C5a are controlled by carboxypeptidase N which converts active C5a into less active C5adesarg.
C5 and Disease
Septicemia is a severe gram-negative bacterial infection of the blood stream and can be complicated by systemic release of anaphylatoxins, namely C5a and C3a, which may cause septic shock and hypotention. The danger of systemic complement activation in the case of sepsis lies in the powerful inflammatory response illicited by activated complement as described above. Anaphylatoxins are beneficial in fighting local infections but may lead to severe illness or death if released on a large scale. Systemic infammatory response syndrome (SIRS) is characterized as a global release of active anaphylatoxin which does not always necessitate bacterial infection (wikipedia.org, 2006). SIRS can lead to septic shock and death.
Systemic lupus erythematosus (SLE) known as lupus is an autoimmune disease of the internal organs. As an autoimmune disease, by definition is an immune response against the host and invovles the complement system. SLE is more likely to occur in individuals with low levels of complement, yet complement is utilized in SLE pathogenesis (Turgeon et al., 2003). The deficiency of complement associated with SLE-infection is due to genetic mutation at a specific complement locus, likely near the locus for C5 (chromosome 9q33). These mutations result in excess inflammation (C5a, C3a, C4a) and cell lysis (MAC, C5b). One study shows that SLE manifestation is reduced when nephritic C5a-R are blocked (Bao et al., 2005).
Leiner's disease is a form of diaper dermatitis seen in newborns and infants. It is characterized by severe generalized seborrhoeic dermatitis, recurrent diarrhea, recurrent skin and internal infections, and failure to thrive. Leiner's diease is not well understood, but it is thought to develop as an inherited form of disfunctional C5 protein (Ngan, 2005).
Liver fibrosis is scarring of the liver tissue and is usually associated with severe liver disease. The involvement of C5 with liver fibrogenesis is not well-characterized, but one study suggests a casual role of the C5 gene and liver fibrosis. Using a murine model, the C5a receptor was inhibited in liver cells of hepatitis C infected individuals which showed antifibrotic effects in vivo. Because the C5 gene is conserved among many organisms, this relationship between C5a and liver fibrogenesis likely holds true for humans (Hillebrandt et al., 2005).
References
Bao L, Osawe I, Puri T, Lambris JD, Haas M, Quigg RJ. 2005. C5a promotes development of experimental lupus nephritis which can be blocked with a specific receptor antagonist. [abstract]. In Eur J Immunology 35(8):2496-506.
Bramham J., Thai C., Soares D., Uhrin D., Ogata R., Barlow P. 2005. Functional Insights from the Structure of the Multifunctional C345C Domain of C5 Complement. [Abstract] Journal of Biological Chemistry. 280:10636-10645 [Abstract] Accessed on 2006 March 16.
Cann A. 2004 October 21. Biological Effects of Complement. www-micro.msb.le.ac.uk/AJC/index.html . Accessed 2006 March 16.
Dahinden CA. 1998. Anaphylatoxins. Encyclopedia of Immunology, Second Edition. Delves PJ. ed. Boston: Academic Press, 1:86-90.
Dalmasso AP. 1998. Complement, Membrane Attack Pathway. Encyclopedia of Immunology, Second Edition. Delves PJ. ed. Boston: Academic Press, 1:624-628.
Hillebrandt S, Wasmuth HE, Weiskirchen R, Hellerbrand C, Keppeler H, Werth A, Schirin-Sokhan R, Wilkens G, Geier A, Lorenzen J, Khol J, Gressner AM, Matern S, Lammert F. 2005. Complement factor 5 is a quantitative trait gene that modifies liver fibrogenesis in mice and humans. [abstract]. Nat Genet. 37(8): 835-43. [Abstract]Accessed on 2006 March 16.
Janeway CA, Traver P, Walport M, Shlomchik MJ. 2005. Immunobiology. 6th Edition. New York: Garland Science.
Ngan, Vanessa 2005. Leiner's Disease. Full Text. Accessed on 2006 March 16.
Sandoval A., Ai R., Ostresh J., Ogata R. 2000. Distal Recognition Site for Classical Pathway Convertase Located in the C345C/Netrin Module of Complement Component C5. The Journal of Immunology. 165:1066-1073.
Stevens, Christine Dorresteyn et al. Clinical Immunology and Serology : a Laboratory Perspective. Philadelphia: F.A. Davis Company, 2003.
Turgeon, Mary Louise et al. Immunology & Serology in Laboratory Medicine, Third Edition . St. Louis: Mosby, 2003.
Wetsel RA, Lemons RS, Le Beau MM, Barnum SR, Noack D, Tack BF. 1991. Complete cDNA sequence of human complement pro-C5. J Immunol. 146 (1): 362-368. Full Text Accessed on 2006 March 14.
wikipedia.org. 2006. Systemic Inflammatory Response Syndrome.SIRS . Accessed on 2006 March 16.
Zhang X, Boyar W, toth MJ, Wennogle L, Connella NC. 1997. Structural definition of the C5a C terminus by two-dimensional nuclear magnetic resonance spectroscopy. [Abstract] Proteins 28:261-7. [Abstract]. Accessed on 2006 March 14.
Questions or comments email: wefiser@davidson.edu