Introduction
Inflammatory bowel disease (IBD) consists of two idiopathic inflammatory diseases ulceritive colitis and Crohn’s disease (CD). The greatest distinction between ulceritive colitis and Crohn’s disease is the range of inflamed bowel tissue. Inflammation in Crohn’s disease is discontinuously segmented known as regional enteritis, while ulceritive colitis is superficial inflammation extending proximally and continuously from the rectum. At present the exact cause of Crohn’s disese is unknown. The disease seems to be related to an exaggerated mucosal immune response to infection of the intestinal epithelium because of an imbalance of proinflammatory and immunoregulatory molecules. The inheritance patterns of Crohn’s disease suggest a complex genetic component of pathogenesis that may consist of several combined genetic mutations. Currently no specific diagnostic test exists for Crohn’s disease, but as understanding of pathogenesis is improved so will testing methods. Treatment of Crohn’s disease consists of inducing remission by anti-inflammatories followed by general immunosuppressants. Emergent therapeutic options focus on specific inflammatory pathways which will halt inflammation and induce remission in patients with Crohn’s disease.
Symptoms
The chronic intestinal inflammation of Crohn’s disease can occur from any point from mouth to rectum but is normally found in the cecum and ileum. The inflammation can spread deep into the bowel tissue causing intestinal ulcers (Carson-DeWitt, 2002). The most common symptom of Crohn’s disease is diarrhea due to increased intestinal secretions of water and salt as well as altered intestinal contractions (Mayo clinic, 2005). Intestinal perforations and fistula formation are seen in some Crohn’s disease patients which may allow intestinal bacteria to invade the abdominal cavity causing further infection. Inflammation may cause thickening and narrowing of the intestinal walls, resulting in bowel obstruction. Other complications include: granulomatosis, hemorrhoids, lipid absorption problems, anemia, anal fissures, artitis, rectal bleeding, and cancer (Mayo clinic, 2005).
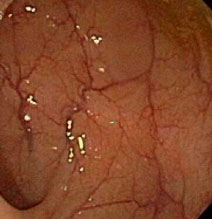
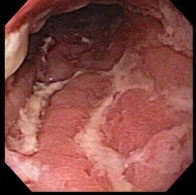
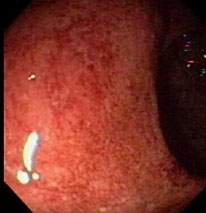
| A. Normal colon |
B. Crohn's disease |
C. Ulceritive Colitis |
| Figure 1. Comparison of normal and inflamed colons by endoscopy. The healthy intestinal tissues (A) show no inflammation. The chronic inflammation in Crohn's disease (B) may lead to complications such as ulcer and fistula formation. Ulceritive colitis (C) is another form of IBD showing superficial inflammation of the intestinal tissues. (images from Achkar, 2006, permission pending). |
Etiology
Autoimmunity
The exact cause of Crohn’s disease is not certain. The disease is associated with a TH1 immune response which results from a loss of tolerance to luminal bacterial antigens. An initial viral or bacterial infection is normally cleared by the mucosal immune system. Antigens cross the epithelium through M cells and are phagocytosed by macrophages which present antigens to CD4+ T cells. Macrophages become activated by bacterial lipopolysaccharides, cytokines, leukotrienes, and prostaglandins which are present after pathogenic infection. Temporary inflammation is usually acquired locally but is controlled, to some extent, by regulatory lymphocytes such as TH3, T regulatory 1, and CD4+/CD25+ T cells (Brookes et al., 2004). Crohn’s disease patients show suppression of these regulatory lymphocytes and an increased production of TH1-associated inflammatory cytokines such as TNF-α, INF-γ, IL-12, and IL-18 (Brookes et al., 2004 and Wirtz et al., 2003). Figure 2 diagrams the proinflammatory and anti-inflammatory pathways of the mucosal immune system. The proinflammatory cytokines cause inflammation by upregulating expression of adhesion molecules such as ICAM-1 and VCAM-1. The increased expression of adhesion molecules recruit more lymphocytes to the infected tissue, resulting in tissue inflammation. In inflammatory bowel disease the constant infiltration of large numbers of macrophages, monocytes, lymphocytes, and plasma cells causes acute inflammation and tissue damage. Tissue damage may occur from prolonged exposure to oxygen radicals and proteases produced by activated leukocytes. Prostaglandins which are synthesized from arachidonic acid and cyclooxygenase cause edema, vasodilation, and pain, and they are found in elevated levels in the stool, blood, and rectal mucosa of Crohn’s patients (Fusunyan et al., 1998).
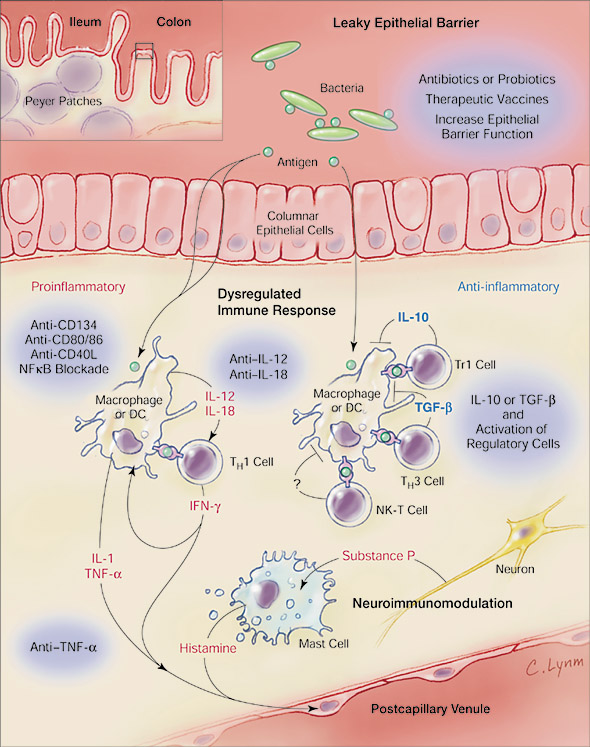
Figure 2. Crohn's disease is primarily characterized by dysfunctional regulatory T cells which normally control proinflammatory cytokine release. If activated antigen presenting cells and lymphocytes are allowed to overly secrete proinflammatory cytokines, then tissue damage may result from prolonged inflammation. (image from Blumberg, 2001, permission pending) |
Many concurrent animal studies have provided clear evidence that balance is achieved in the mucosal immune system by regulatory cytokines which both stimulate apoptosis in activated immune cells and inhibit proinflammatory cytokine release as well as antigen presentation (Wirtz et al., 2003). Crohn’s disease is considered to be a condition whereby regulatory lymphocytes and cytokines fail to control the mucosal immune response resulting in prolonged inflammation and intestinal tissue damage. The result of differential cytokine expression and deregulation of inflammatory molecules directs other branches of the immune system to deviate from the healthy norm.
Another factor contributing to intestinal inflammation is a change in immunoglobulin isotype secretions. Normal intestinal tissues contain high levels of IgA, which functions as a pathogen neutralizer in intestinal tissue. Patients with Crohn’s disease show an elevated level, nearly 30 times the norm, of IgG-producing plasma cells which secrete predominantly IgG1 and IgG2. IgG plays a role in inducing inflammation by fixing complement, arming NK cells, and activating phagocytes (Fusunyan et al., 1998).
In IBD and colonic diverticulitis activated T cells and macrophages readily shed soluble IL-2 receptor molecules. Increased serum IL-2 receptor levels in Crohn’s disease patients correlate with intestinal inflammatory activity as IL-2 receptor levels are detected in intestinal venous tissue of Crohn’s patients (not in ulceritive colitis) (Fusunyan et al., 1998). Since IL-2 and its receptor are directly involved with lymphocyte activation and proliferation, the relative level of IL-2 receptor in circulation provides a useful marker of disease progression (Fusunyan et al., 1998).
Genetic susceptibility
Because about 20% of Crohn’s patients have a parent, sibling, or child with the disease, a strong genetic component to disease susceptiblity is suggested (Mayo clinic, 2005). At risk populations include Ashkenazi Jews of European decent who are four to five times more likely to have Crohn’s disease and Caucasians with an incidence of 2-20 per 100,000 individuals (Carson-DeWitt, 2002). Also the following observations support the genetic basis of Crohn’s disease:
1 – Increased monozygotic twin concordance rates (50% in Crohn’s) compared to dizygotic twins 2 – Rarity of IBD in spouses 3 – Disease seen in relatives independent of geophraphic or temporal differences. (Yang et al., 1997)
Genetic observations of Crohn’s disease populations show that the disease does not follow simple Mendelian inheritance patterns. Rather, IBD is thought to be several etiologically and genetically distinct diseases showing clinical similarity. Support for this theory comes from murine studies in which genes for IL-2, IL-10, or T cell receptor were disrupted, and intestinal lesions similar to human IBD were noted (Yang et al., 1997).
Identifying causitive genes for Crohn’s disease is difficult because of its etiologically heterogeneous nature and non-Mendelian inheritance characteristics. However, candidate genes, those which are involved in the immunological pathway of Crohn’s disease, have been targeted for study. The most actively investigated gene is the HLA region (HLA-DR, HLA-DQ, and HLA-DP) on chromosome 6 which codes for the major histocompatibility complex (MHC) (Yang et al. 1997). It was observed that HLA haplotype is shared among siblings with Crohn’s disease. The effect of HLA mutation on disease onset is modest, but it is a contributing factor, nonetheless. “It is very intriguing that the genetic markers in the region with the greatest relative risk for CD are genetic variations surrounding the TNF genes” (Yang et al., 1997). This observation further supports evidence that TNF-α plays an important role in Crohn's disease. Another candidate gene is the single base change polymorphism codon 241 of the ICAM-1 binding domain. Although initial studies showed low correlation between codon 241 of ICAM-1 and Crohn’s disease, this polymorphic gene segment might be used to identify genetically heterogeneous subsets of Crohn’s disease (Yang et al., 1997).
Another suspected genetic contributor is NOD2 (CARD15) which is an intracellular protein in macrophages having a caspase-recruitment domain (CARD). The same CARD is found in proteins that execute apoptosis by activating the NFkB pathway. NOD2 protein has a leucine-rich repeat domain similar to the Toll-like receptor, suggesting NOD2 may act as an intracellular recpetor for bacteria. Mutations in NOD2 are highly associated with IBD and may cause disease by initiating an abnormal response to gut flora or failure to induce apoptosis in activated macrophages (Janeway et al., 2005 and Brookes et al., 2004).
Diagnostics
No diagnostic has been developed with complete specificity for IBD. Therefore, other tests are performed which may rule out other diseases having similar clinical presentation. Colonoscopy exams are used to identifiy granulomas which are a distinction of Crohn’s disease. Other diagnostic tools include: sigmoidoscopy, barium enema, small bowel X-ray, CT scan, and capsule endoscopy (Mayo clinic, 2005). The Crohn’s Disease Activity Index is one of the most widely accepted assessments of disease progression. This index assigns a numerical rating (150-450) based on acuity of clinical presentation and patient response (Brookes et al., 2004)
Therapy
Current therapeutic options
The treatment for Crohn’s disease is aimed at reducing inflammation with anti-inflammatory drugs and immuno-supporessants. Abcesses may require antibiotics to clear bacterial infection. The last option for treating Crohn’s disease is colectomy, and 60% of Crohn’s patients will require surgery (Carson-DeWitt, 2002). The main anti-inflammatories used for Crohn’s disease therapy are sulfasalazine, mesalamine, and corticosteroids (Mayo clinic, 2005). Sufasalazine contains salicylic acid and antibiotics, giving it dual action to reduce inflammation and fight bacteria. Corticosteriods inhibit inflammation anywhere in the body by reducing the proinflammatory cytokine INF-γ which is secreted by activated T cells. Corticosteroids bind to the AP-1 active site on INF-γ promoter gene segment and suppress transcription (Barbulescu et al., 1998) The utility of corticosteriods in IBD therapy is that they effectively induce remission so that immunosuppressants can maintain remission. Corticosteroids should not be used for long periods of time as they may cause hypertension, adrenal insufficiency, and dependency (Brookes et al., 2004).
Introduced in 1998 as the first biological agent for Crohn’s disease, Infliximab is a chimeric (75% human, 25% murine) IgG1 monoclonal antibody for TNF-α and is an immunosuppressant widely used in Crohn’s disease therapy. Infliximab binds to both soluble and membrane bound TNF-α, thereby neutralizing the proinflammatory cytokine and reducing inflammation (Brookes et al., 2004). In one study 81% of Crohn’s patients responded to Infliximab and 62% of patients maintained remission (Brookes et al., 2004). Infliximab also acts by downregulating CD40 and VCAM-1 expression in intestinal microvessels (Danese et al., 2006). CD40 and its ligand stimulate the production of TNF-α which acts as a positive feedback loop by increasing CD40 and CD40L expression. VCAM-1 is normally absent in intestinal tissue but is increased significantly in Crohn’s disease. By decreasing VCAM-1 expression with Infliximab, fewer immune cells can enter the tissue which reduces inflammation (Danese et al., 2006). Interestingly, patients with Crohn’s disease and latent tuberculosis sometimes experience activation of tuberculosis (Brookes et al., 2004 and Mayo clinic, 2005). Since TNF-α is a TH1-mediated cytokine and TH1 cells control mycobacterial infections, inhibition of TNF-α should correlate with proliferation of mycobacteria.
Emerging therapeutic options
Current therapeutic research is targeted towards normalizing the altered immune response in IBD. More therapeutic options will become available as the immunologic etiology is further illucidated. Etanercept is a soluble TNF-α receptor fusion protein which inhibits TNF-α pro-inflammatory action by sequestering TNF-α in circulation. Etanercept is being considered for Crohn’s disease therapy but is not yet recommended for clinical use (Brookes et al., 2004). Another biological agent for therapy is CDP-571 which is a 95% humanized monoclonal IgG4 antibody specific for TNF-α. CDP-571 holds much biological similarity to Infliximab but is less likely to fix complement or induce antibody-dependent cytotoxicity in TNF-α producing cells (Brookes et al., 2004).
Other therapeutic drugs under consideration are recombinant human IL-10 therapy and adhesion molecule inhibitors. The cytokine IL-10 is known to stimulate a TH2 response as well as promote regulatory lymphocytes. A drug which delivered IL-10 to inflammed intestinal tissue would likely induce remission in Crohn’s patients by inhibiting a TH1 response and macrophage activation and thus halting further inflammation (Brookes et al., 2004). Gene transfer therapy for regulatory cytokines such as IL-10 is currently being explored and yields positive preliminary results (Wirtz et al., 2003). Alicaforsen is another immunosuppressant currently being studied which targets the ICAM-1 receptor. It acts by inhibiting the mRNA translocation of the ICAM-1 receptor which is upregulated during inflammation to attract more immune cells to the infected tissue (Brookes et al., 2004). As the immunology and genetics of the inflammatory pathways in IBD are better understood, more effective biological therapy can be developed, providing improved care for those who suffer from Crohn's disease.
References
Achkar JP. Inflammatory Bowel Disease. American College of Gastroenterology. [link] Accessed 2006 April 17.
Barbulescu K, Becker C, Zum Buschenfelde K, Neurath M. 1998. Regulation of protein-DNA interactions at the interferon-gamma gene promoter by corticosteroids. Annals of the New York Academy of Sciences. 859: 319-22.
Blumberg RS and Strober W. 2001. Prospects for research in inflammatory bowel disease. JAMA. 285(5): 643-7.
Brookes MJ and Green JR. 2004. Maintenance of remission in Crohn’s disease: current and emerging therapeutic options. Drugs. 64(10): 1069-89.
Carson-DeWitt RS. 2002. Crohn’s disease. The Gale Encyclopedia of Medicine Second Edition. Longe D. ed. Farmington Hills. 2: 956-959.
Danese S, Sans M, Franco S, Sgambato A, Rutella S, Cittadini A, Pique J, Panes J, Katz J, Gasbarrini A, Fiocchi C. 2006. TNF-α Blockade Down-Regulates he CD40/CD40L Pathway in the Mucosal Microcirculation: A Novel Anti-Inflammatory Mechanism of Infliximab in Crohn’s Disease. Journal of Immunology. 176: 2617-2624.
Fusunyan R and Sanderson I. 1998. Inflammatory Bowel Disease. Encyclopedia of Immunology, Second Edition. Delves PJ. ed. Boston: Academic Press, 3: 1375-1381.
Janeway CA, Traver P, Walport M, Shlomchik MJ. 2005. Immunobiology. 6th Edition. New York : Garland Science.
Mayo Clinic Staff. 2005. Crohn’s disease. MayoClinic.com. [link]. Accessed 2006 April 12.
Wirtz S and Neurath MF. 2003. Gene transfer approaches for the treatment of inflammatory bowel disease. Gene Therapy. 10: 854-860.
Yang H and Rotter J. 1997. Inflammatory Bowel Disease. Encyclopedia of Human Biology. Dulbecco R. ed. San Diego: Academic Press, 5: 59-65.
created by Wesley Fiser 2006, Davidson College
Questions or comments email me wefiser@davidson.edu