-This web page was produced as an assignment for an undergraduate course at Davidson College-
***DISCLAIMER: This website is not intended to be used for medical advice or treatment.***
ADA/SCID
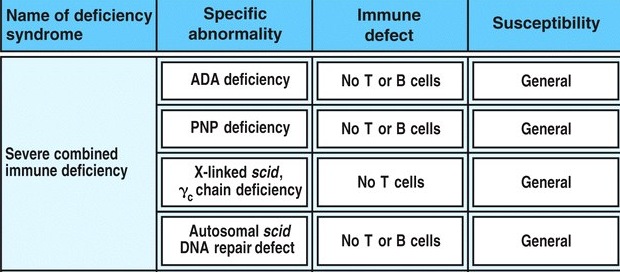
Severe combined immunodeficiency (SCID) is a human disorder characterized by defective T-cell function (Janeway, 2005). All forms of SCID are inherited (Kniffin, 2004). Severe combined immunodeficiency is associated with recurring opportunistic infections and often results in death at a young age (Blaese et al., 1995). There are numerous defects that result in the SCID phenotype and several of the most common causes are listed in Table 1. The most common form of SCID is X-linked SCID, which is caused by a deficiency of the IL2 receptor. Another common cause of SCID is adenosine deaminase (ADA) deficiency; approximately 20% of the SCID conditions diagnosed are the result of ADA deficiency (Carlucci et al., 2004). In fact, adenosine deaminase deficiency is the most common form of autosomally inherited SCID (Janeway et al., 2005).
|
| Table 1 lists several of the common causes of severe combined immune deficiency, as well as the immune defect that they generate and the type of immune susceptibility that affected individuals experience. Fig 11.8 from Janeway et al., 2005. |
Adenosine deaminase deficiency resulting in severe combined immunodeficiency (ADA/SCID) in humans is characterized by a severely compromised immune system and a lack of functional T and B lymphocytes in affected individuals. The enzyme adenosine deaminase functions as a catalyst in the deamination of adenosine and deoxyadenosine. (Valerio et al., 1984). Specifically, ADA catalyzes the irreversible deamination of adenosine and deoxyadenosine to inosine and deoxyinosine (Zavialov and Engstrom, 2005). ADA deficiency results in an accumulation of deoxyadenosine nucleotides, which are toxic to T lymphocytes and inhibit their development and inhibit the development of other lymphoid cells including B and NK cells. Most importantly, these metabolites affect the ability of T cells to differentiate in mature T cells (Janeway et al., 2005). Two distinct isoenzymes of adenosine deaminase, ADA1 and ADA2, have been identified in human beings. Mutations in the human ADA1 gene lead to inherited SCID as ADA1 is responsible for most the adenosine deaminase activity found in humans (Zavialov and Engstrom, 2005). The ADA1 gene, which is located on chromosome 20, contains 12 exons and spans 32 kilobases, and the isolated cDNA encodes for a 363-amino acid protein (Kniffin, 2004).
Recent research has shed considerable light on the mechanisms of ADA deficiency that result in SCID. The toxicity of accumulated adenosine and deoxyadenosine is mediated by both intra- and extracellular pathways. Normally, adenosine is believed to function in T cell differentiation via signaling through adenosine receptors. When adenosine accumulates in T cells in the absence of adenosine deaminase, it leads to formation of toxic levels of deoxyadenosine and deoxyATP. Recent research has shown that while this intracellular mechanism significantly contributes to the death of many developing lymphocytes, extracellular mechanisms of ADA deficiency may contribute to the ADA/SCID condition as well. Extracellular adenosine interferes with thymocyte selection by interacting with adenosine receptors and inhibiting the signaling that occurs through T cell receptors (TCR). Therefore, those lymphocytes that do survive the toxic effects of intracellular adenosine may not survive positive selection, which relies on TCRs. Furthermore, the extracellular effects of adenosine may contribute to selection of T cells with a high TCR avidity and thus create an imbalance between effector and regulatory cells in the surviving lymphocyte population. This theory can help explain the autoimmunity observed in many patients suffering from ADA/SCID (Apasov and Sitkovsky, 1999).
The ultimate cause of ADA/SCID has been the subject of intense research. In individuals with ADA/SCID, the defect does not lie in the expression of the ADA gene; rather, the ADA protein created by ADA/SCID patients exhibits an altered conformation, affecting its catalytic function and stability. Thus, the ultimate cause of ADA deficiency is not a lack of ADA protein, but a lack of functional ADA protein (Valerio et al., 1984). The lack of functional ADA protein leads to an accumulation of the ADA substrate deoxyadenosine, which is converted to deoxyadenosine triphosphate in T cells with toxic effects (Blaese et al., 1995). The structure of murine ADA can be seen in Figure 1.
| Figure 1 depicts the structure of murine adenosine deaminase as rendered by Jmol. Protein structure acquired from the Protein Data Bank. |
This condition holds an important place in science and medicine; ADA/SCID was the first disease treated with gene therapy. Because it is the result of a defect in only one gene, ADA/SCID was an attractive candidate for early gene therapy trials (Carlucci et al., 2004). The first gene therapy trials performed on patients with ADA/SCID were done in concert with bovine enzyme replacement therapy and polyethylene glycol (PEG) (Aiuti et al., 2002 2). The addition of PEG to bovine ADA prevents clearance of the enzyme from circulation and prolongs the half-life of ADA in plasma (Bordignon et al., 1995). The treatment, known as PEG-ADA, is expensive and does not cure SCID, but rather provides patients with a source of ADA to prevent toxic buildup of deoxyadenosine. The use of PEG-ADA allows for the survival of T lymphocytes, which provided the source of cells used in the gene therapy trials (Blaese et al., 1995).
The first successful gene therapy trial was performed in 1990 on two infant girls with ADA/SCID (Anderson, 2002). Prior to gene therapy, PEG-ADA therapy and bone marrow transplant were the only options for treatment of ADA/SCID. Doctors could not justify taking patients off of PEG-ADA therapy due to the demonstrated benefits of the treatment. Therefore, the successful results of initial gene therapy trials could not be fully attributed to gene therapy. However, in 2002, doctors identified one patient in which the discontinuation of bovine enzyme therapy could provide clear benefits. The patient, who had been treated with gene-corrected peripheral blood lymphocytes (PBLs), exhibited significant expansion of gene-corrected T lymphocytes, as these cells enjoyed a selective advantage in the absence of PEG-ADA. Furthermore, these patients showed improved immune function and were able to form responses to T cell-dependent antigens such as tetanus toxoid. Thus, at least in the case of PBL gene therapy, it appears as though gene therapy alone can produce a competent immune system in ADA/SCID individuals (Aiuti et al., 2002 2). Gene therapy on ADA/SCID patients may now be performed without the use of PEG-ADA therapy (Aiuti et al., 2002 1).
Hematopoietic stem cells (HSCs) have been the target of subsequent gene therapy trials for ADA/SCID, but have not enjoyed the same success in providing immunity as the earlier T-cell trials. This lower success rate has been blamed on the poor efficiency observed during the transfer of genes into HSCs. Improvements in the gene transfer protocol for HSC gene therapy include nonmyeloblative conditioning of the patients following transfer of gene-corrected HSCs, which provides an advantage to the transduced cells and creates space in the bone marrow (Aiuti et al., 2002 1). Further trials have demonstrated that long-term immunity in ADA/SCID patients can be achieved using gene therapy directed towards bone marrow derived stem cells (Bordignon et al., 1995).
For several decades prior to the beginning of gene therapy, scientists and researchers were able to introduce human genes into non-human hosts. Human gene therapy first began in 1990 and since then more than 4,000 people have been treated. Gene therapy is defined as the introduction of genetic material into a patient’s own genome. The end goal of gene therapy is the replacement of a non-functioning or mutant allele with the correct gene, conferring normal protein function in the host. The first step of gene therapy involves the identification and isolation of the gene of interest. After the gene has been cloned, the cDNA is inserted into a vector to produce a chimera. A wide range of molecules may be used as vectors but in the case of SCID retroviruses are most commonly utilized. Retroviruses use their viral machinery to insert their genome into the host cell's DNA, and can be engineered to carry the target genes for gene therapy. The gene/vector hybrid or chimera must then be transfected into the host either in vivo or ex vivo. ADA/SCID gene therapy trials have used ex vivo transfection; cells are removed from the host, transfected by the chimera, grown in culture for 9 to 12 days, and then returned to the host. The resulting organism is said to be transgenic (Simon, 2002).
In the first gene therapy trial, which was performed at the National Institutes of Health, a retroviral vector was used to insert 1.5 kilobase cDNA into the mature T lymphocytes of two young girls (Simon, 2002). The vector used in the first gene therapy trials was a LASN retrovirus (Blaese et al., 1995). Doctors treated the girls for a total of eleven times over two years. The girls, who were not candidates for a bone marrow transplant, were also receiving PEG-ADA therapy at a cost of $400,000 per year. To the great excitement of the medical community, both experienced a significant improvement in immune function and were able to leave quarantine and lead fairly normal lives (Simon, 2002).
Since the initial trials for ADA/SCID, other types of SCID have been treated with gene therapy. The most notable of these is X-linked SCID. X-linked SCID can be treated by transplant of hematopoietic stem cells from bone marrow, but the difficulty of matching donors with recipients and the risks of graft rejecting have made gene therapy an attractive treatment option for the condition. The French team, led by Drs. Alain Fischer and Marina Cavazzanna-Calvo, began the trial at the Necker Hospital for Sick Children in Paris, France. X-linked SCID is often known as the “bubble baby” disease as the result of the sterile housing in which affected infants must be kept (ASGT, 2003). X-linked SCID is also known as IL2RG deficiency; patients with the condition were treated with a retrovirus containing the IL2RG gene (Dave and Jenkins, 2004). In the French trial, doctors inserted the corrective gene into hematopoietic stem cells from the patient’s own bone marrow; these cells then differentiated into T and B cell lymphocytes capable of launching an effective immune response (ASGT, 2003). The French trial made use of a Moloney-derived retroviral vector (MFG), which has been demonstrated to insert more efficiently the vectors used in early trials (Anderson, 2000).
The French trial initially appeared to have been a success; using a retrovirus to insert corrective genes into blood stem cells, the French team had restored sufficient immune function in nine of ten children with X-linked SCID, which is normally a fatal condition without treatment (FDA, 2003). In fact, gene therapy pioneer W. French Anderson had cited the French doctors’ success in correcting the X-linked SCID disorder with stem cell gene therapy (2002). However, two patients in the French trial developed T cell leukemia within 30 months (Dave and Jenkins, 2004).
The revelation of the multiple cancers occurring in the French trial raised serious concerns regarding the safety of gene therapy. On January 14, 2003, the U.S. Food and Drug Administration announced that it would be temporarily halting all gene therapy trials in the United States in the wake of the announcement that a second child in the French gene therapy trial for X-linked SCID had developed leukemia. Previously, in August of 2002, the doctors performing the trial had announced one the ten children in the study had developed a leukemia-like condition. At that time, the FDA responded by stopping enrollment of patients in U.S. gene therapy trials that resembled the French trial (FDA, 2003).
Prior to this development there had been previous warnings about the potential dangers surrounding gene therapy. The first major setback in gene therapy occurred on September 17, 1999 when an 18-year old male died while undergoing gene therapy at the University of Pennsylvania Medical Center. Doctors later determined that the man had died from a systemic immune response to the adenovirus vector used in the trial. Trials continued in the wake of the tragedy, but with heightened scrutiny from the government and medical community (Simon, 2002).
The American Society of Gene Therapy supported the FDA’s temporary halt of gene therapy trials in the U.S. but emphasized the potential of gene therapy in the treatment of genetic disorders. The development of cancer has been acknowledged as one of the potential detrimental outcomes of gene therapy; the use of retroviruses to insert genetic material may contribute to cancer if the gene and its promoter are inserted in the vicinity of an oncogene (ASGT, 2003). Researchers raced to discover why there appeared to be a heightened risk of cancer in the X-linked SCID trials. They were able to determine that the retrovirus inserted in the vicinity of LMO2, which is classified as a T cell oncogene. Previously, this insertion had been assumed to be completely random, and thus the chance of activating an oncogene was believed to be sufficiently small compared to the potential for correcting an otherwise fatal condition. However, the devastating results of the trial raised the concern that insertion may be non-random; in fact, this was shown to be the case for the particular gene, and researchers have suggested that all the patients in the trial received an insertion at the LMO2 gene. The inserted gene IL2RG is believed to act as an oncogene when expressed by a retrovirus, and when working in concert with LMO2, may lead to the expansion of the host cell into leukemia. This finding of course raises the question of why all of the children did not develop leukemia; researchers suspect that other subtle genomic mutations may have provided the spark for the cancer in the two affected patients (Dave and Jenkins, 2004).
In all, a total of three children in Dr. Fischer’s trial at the Necker Hospital developed a leukemia-like condition. One of the ten children in the trial died in fall 2004. While the first two diagnosed cases involved the LMO2 oncogene, the third case appears to involve a different oncogene and insertion occurred at three different sites. Thus far, no cases of cancer have been observed in gene therapy trials for ADA/SCID, and a government oversight panel suggested in 2005 that the FDA lift its hold in gene therapy trials for ADA/SCID, but suggested that trials for X-linked SCID remain restricted (Kaiser, 2005). Indeed, gene therapy is a tool that merits considerable discussion and review because of both the potential benefits and risks.
If available, click on the ![]() to view the PubMed abstract
to view the PubMed abstract
![]() Aiuti, A, S Vai, A Mortellaro, G Casorati, F Ficara, G Andolfi, G Ferrari, A Tabucchi, F Carlucci, HD Ochs, LD Notarangelo, MG Roncarolo, C Bordignon. (2002). Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nature Medicine. 8: 423-5.
Aiuti, A, S Vai, A Mortellaro, G Casorati, F Ficara, G Andolfi, G Ferrari, A Tabucchi, F Carlucci, HD Ochs, LD Notarangelo, MG Roncarolo, C Bordignon. (2002). Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nature Medicine. 8: 423-5.
![]() Aiuti, A, S Slavin, M Aker, F Ficara, S Denla, A Mortellaro, S Morecki, A Shoshana, T Grazia, C Antonella, F Carlucci, E Marinello, F Cattenio, V Federica, S Sergion, M Paolo, R Miniero, M Roncarolo, B Grazia, C Bordignon. (2002). Correction of ADA-SCID by stem cell gene therapy, combined with nonmyeloablative conditioning. Science. 296: 2410-13.
Aiuti, A, S Slavin, M Aker, F Ficara, S Denla, A Mortellaro, S Morecki, A Shoshana, T Grazia, C Antonella, F Carlucci, E Marinello, F Cattenio, V Federica, S Sergion, M Paolo, R Miniero, M Roncarolo, B Grazia, C Bordignon. (2002). Correction of ADA-SCID by stem cell gene therapy, combined with nonmyeloablative conditioning. Science. 296: 2410-13.
![]() Apasov, SG, MV Sitkovsky. (1999). The extracellular versus intracellular mechanisms of inhibition of TCR-triggered activation of thymocytes by adenosine under conditions of inhibited adenosine deaminase. International Immunology. 11: 179-89.
Apasov, SG, MV Sitkovsky. (1999). The extracellular versus intracellular mechanisms of inhibition of TCR-triggered activation of thymocytes by adenosine under conditions of inhibited adenosine deaminase. International Immunology. 11: 179-89.
American Society of Gene Therapy. 2003 Jan 14. American Society of Gene Therapy responds to a second case of Leukemia seen in clinical trial of gene therapy for immune deficiency. News Release. http://www.asgt.org/news_releases/01132003.html. Accessed 2006 Apr 19.
Anderson , WF. (2000). The best of times, the worst of times. Science. 288: 627-8.
![]() Blaese, RM, KW Culver, AD Miller, CS Carter, T Fleisher, M Clerici, G Shearer, L Chang, Y Chiang, P Tolstoshev, JJ Greenblatt, SA Rosenberg, H Klein, M Berger, CA Mullen, WJ Ramsey, L Muul, RA Morgan, WF Anderson. (1995). T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science. 270: 475-80.
Blaese, RM, KW Culver, AD Miller, CS Carter, T Fleisher, M Clerici, G Shearer, L Chang, Y Chiang, P Tolstoshev, JJ Greenblatt, SA Rosenberg, H Klein, M Berger, CA Mullen, WJ Ramsey, L Muul, RA Morgan, WF Anderson. (1995). T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science. 270: 475-80.
Bordignon, C, LD Notarangelo, N Nobili, G Ferrari, G Casorati, P Panina, E Mazzolari, D Maggioni, C Rossi, P Servida, AG Ugazio, F Mavilio. (1995). Gene therapy in peripheral blood lymphocytes and bone marrow for ADA/SCID immunodeficient patients. Science. 270: 470-5.
![]() Carlucci, F, A Tabucchi, A Aiuti, F Rosi, F Floccari, R Pagani, E Marinello. (2004). Evaluation of ADA gene expression and transduction efficiency in ADA/SCID patients undergoing gene therapy. Nucleosides, Nucleotides, & Nucleic Acids. 12: 1245-48.
Carlucci, F, A Tabucchi, A Aiuti, F Rosi, F Floccari, R Pagani, E Marinello. (2004). Evaluation of ADA gene expression and transduction efficiency in ADA/SCID patients undergoing gene therapy. Nucleosides, Nucleotides, & Nucleic Acids. 12: 1245-48.
Dave, UP, NA Jenkins, NC Copeland. (2004). Gene therapy insertional mutagenesis insights. Science. 303: 333.
![]() Kaiser, J. (2005). Panel urges limits on X-SCID trials. Science. 307: 1544-5.
Kaiser, J. (2005). Panel urges limits on X-SCID trials. Science. 307: 1544-5.
Kniffin, CL. (2004). Adenosine deaminase; ADA. Online Mendelian Inheritance in Man, John Hopkins University. http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=608958. Accessed 2006 22 Apr.
Janeway, CA, P Travers, M Walport, MJ Shlomchik. Immunobiology 6. NewYork: Garland Publishing, 2005.
Simon, EJ. (2002). Human gene therapy: genes without frontiers? The American Biology Teacher. 64: 264-70.
U.S. Food and Drug Administration. 2003 Jan 14. FDA places temporary halt on gene therapy trials using retroviral vectors in blood stem cells. FDA Talk Paper. http://www.fda.gov/bbs/topics/ANSWERS/2003/ANS01190.html. Accessed 2006 Apr 19.
![]() Valerio, D, MGC Duyvesteyn, HV Ormondt, PM Khan, AJ van der Eb. (1984). Adenosine deaminase ( ADA) deficiency in cells derived from humans with severe combined immunodeficiency is due to an aberration of the ADA protein. Nucleic Acids Research. 12: 1015-24.
Valerio, D, MGC Duyvesteyn, HV Ormondt, PM Khan, AJ van der Eb. (1984). Adenosine deaminase ( ADA) deficiency in cells derived from humans with severe combined immunodeficiency is due to an aberration of the ADA protein. Nucleic Acids Research. 12: 1015-24.
![]() Zavialov, AV, A Engstrom. (2005). Human ADA2 belongs to a new family of growth factors with adenosine deaminase activity. Biochemical Journal. 391: 51-7.
Zavialov, AV, A Engstrom. (2005). Human ADA2 belongs to a new family of growth factors with adenosine deaminase activity. Biochemical Journal. 391: 51-7.
This page created by: Kyle Kinsell
Return to the Immunology Homepage
Return to Kyle Kinsell's Immunology Homepage
![]()
![]()