This web page created as an assignment for an undergraduate course.
*
Please consult a physician if you need medical advice.
Overview
Genetic Risk Factors
Pathology
Symptoms
Diagnosis
Treatment
Interview: Living with Celiac Disease
Links and Resources
References
In literature, this disease is alternatively referred to as: coeliac disease, celiac sprue, gluten-sensitive enteropathy, nontropical sprue, and gluten intolerance.
Celiac disease is a hereditary intolerance to gluten, a protein found in wheats and, to a lesser extent in barley, rye, and oats. While gluten intolerance is permanent, symptoms can be alleviated by avoiding all gluten from one’s diet. Inflammation occurs when gliadin, a peptide derivative of gluten, found in gluten-containing foods is ingested and presented to T cells. Inflammation causes damage to mucosal tissue of the small intestine, especially the villi that absorb nutrients, which results in malabsoption of food. (Westerberg, et al., 2006). Celiac disease affects as many as 1 in 300 people in Italy and southwestern Ireland, but is extremely rare in Africa, Japan, and China ( Not et al., 1988). According to a multicenter study in 2003, there is a 1 in 133 chance that people with no risk factors or family history in the U.S. have celiac disease. Additionally, a person’s risk increases to a 1 in 22 chance if they have a first-degree relative with celiac disease and a 1 in 39 chance if they have a second-degree relative (Fasano, 2002). Around 60,000 Americans are diagnosed with celiac disease annually and a total of over 2 million have the disease, making it perhaps the most common genetic disorder in the United States (Westerberg, et al., 2006). Celiac disease can occur at any age, and females are more commonly affected than males. Of females presenting during their fertile years, the male to female ratio is almost 3 to 1 (Feighery et al., 1998).
Celiac disease is strongly associated with the human leukocyte antigen (HLA) DQ2 and DQ8 haplotypes. HLA genes are part of the major histocompatibility complex (MHC). The function of MHC molecules is to bind peptide fragments derived from pathogens and display them on the cell surface for recognition by T cells. Many proteins involved in antigen processing and presentation are encoded by genes within the MHC (Janeway, 2005).
Symptoms of celiac disease are caused by a glutamine and proline rich 33-mer peptide found in gluten that initiates the inflammatory response when bound to these HLA haplotypes (Shan, Lu et al, 2002). The primary HLA association in most patients with celiac disease is with DQ2 (DQA*05/DQB1*02) and in a minority of patients with DQ8 (DQA1*03/DQB1*0302). Approximately 97% of individuals with celiac disease have the HLA-D2Q or HLA-DQ8, compared to 40% of the general population (NIH, 2006). The x-ray crystal structure of the soluble domain of HLA-DQ2 was observed bound to the deaminated gluten epitope alpha-I-gliadin. The HLA association in celiac disease can be explained by a superior ability of DQ2 to bind the repertoire of proline-rich gluten peptides that have survived gastrointestinal digestion and that have been deaminated by tissue transglutaminase (Kim et al., 2004).
Celiac disease is diagnosed in about 10% of first degree relatives of an individual with celiac disease (Logan, 1992). Although hereditary factors play a significant role, genetic factors alone do not explain the development of the disease because the disease is concordant in only 60% to 70% of identical twins. All people with HLA DQ2 and DQ8 do not develop into disease, and others without those alleles can develop celiac disease, so more genes may be involved. Additional factors such as hormones and infectious agents may also be involved in linking the ingestion of gluten with a chronic inflammatory reaction in the intestine in genetically predisposed individuals (Fasano,1996).
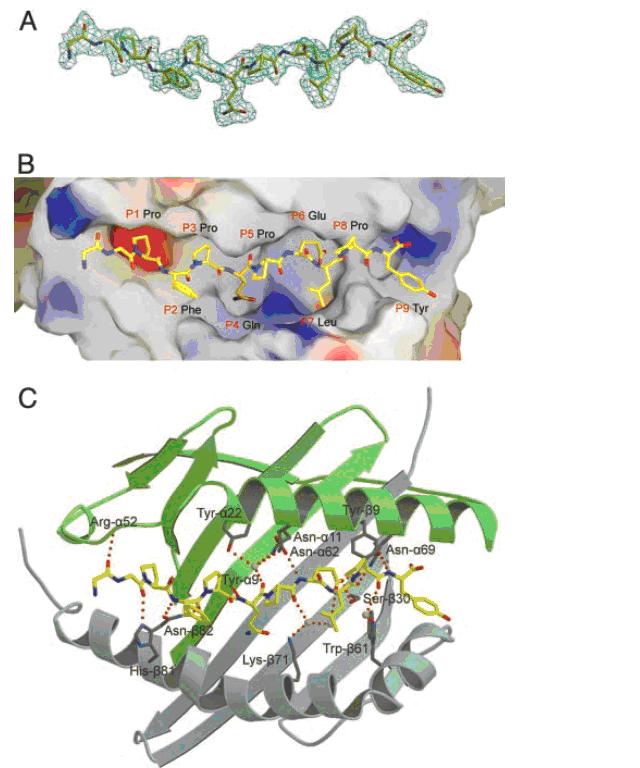
Figure 1: Gliadin peptide fragment derived from gluten protein is presented by antigen-presenting cells with specific class II HLA-DQ alleles.
(A)Model of the gliadin peptide. (B) Surface of HLA-DQ with bound gliadin peptide to red region. (C) Hydrogen-bonding network in the HLA-DQ-gliadin complex (image from Kim, C. et al., 2004, permission pending).
Innate and adaptive immune responses are involved in celiac disease. Recent studies show the importance that toll-like receptors (TLR) of the innate immune system have in intestinal homeostasis. Ingestion of gliadin-containing grains initiates TLR signaling through myeloid differentiation factor 88 (MyD88), a key adapter molecule in the TLR/IL-1R pathway. Peptide derivatives of gliadin stimulate the MyD88-dependent release of zonulin which increases intestinal permeability. This enables paracelluar translocation of gliadin and its subsequent interaction with macrophages in the intestinal submucosa. The interaction of gliadin with macrophages causes a release of MyD88-dependent proinflammatory cytokines which facilitates the interaction of T cells with antigen-presenting cells (Thomas, 2006). The enzyme tissue transglutaminase (tTG) also has an important role in modifying gluten-derived peptides into glutamic acid which can bind to HLA-DQ2/8 molecules, since these HLA molecules preferentially bind to peptides with negative charges in specific anchor residues ( Papadopoulos et al., 2001).
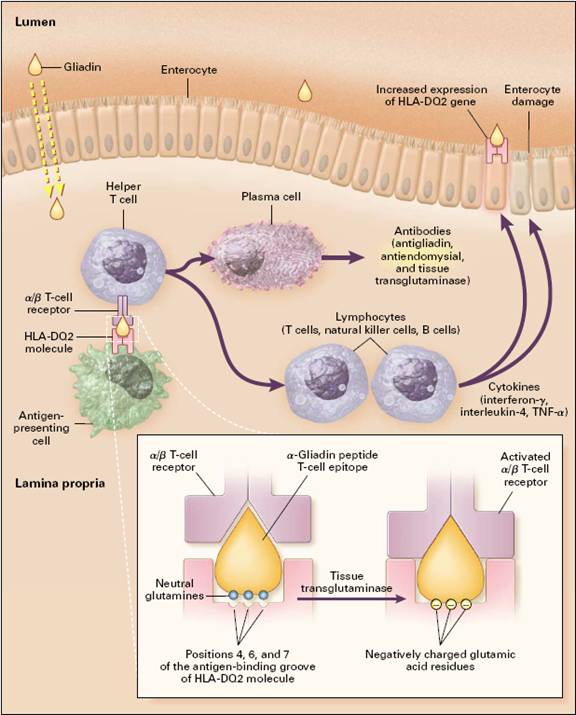
Figure 2: Pathogenesis of Celiac Sprue
(figure from Westerberg et al., 2006 permission pending)
Gliadin is absorbed into the lamina propia and presented in conjunction with HLA-DQ2 or DQ8 cell-surface antigens by antigen-presenting cells, probably dendritic cells, to sensitized T cells expressing the α /β T-cell receptor. Tissue transglutaminase deaminates gliadin peptides, generating acidic, negatively charged residues of glutamic acid from neutral glutamines (inset). Since negatively charged residues are preferred in positions 4,6, and 7 of the antigen-binding groove of HLA-DQ2, deaminated gliadin elicits a stronger T-cell response. These lymphocytes then activate other lymphocytes to generate cytokines, such as interferon-γ, interleukin-4, and tumor necrosis factor α (TNF- α), which damage the villi, resulting in enteritis. Induction of aberrant HLA class II cell-surface antigens on the enterocytes may permit these cells to present additional antigens to the sensitized lymphocytes (Westerberg et al.,2006).
Celiac disease symptoms may start in childhood or adulthood, with onset and severity influenced by the amount of gluten that is eaten. These autoantibodies cause inflammation in and damage to the lining of the intestinal wall. Eventually, decreased absorption of nutrients (malabsorption) can cause vitamin deficiencies that deprive the brain, peripheral nervous system, bones, liver and other organs of vital nourishment, which can lead to other illnesses (Mayo Clinic, 2006). This causes symptoms associated with malnutrition and malabsorption, such as: diarrhea, weakness, weight loss, abdominal pain, abdominal distention, fatigue, oral ulceration, bleeding tendency, bone and joint pain, and anemia. Even in patients with absent or minor gastrointestinal symptoms, celiac disease can manifest itself cutaneously as dermatitis herpetiformis, a skin disorder associated with papulovesicles on the knees, elbows, buttocks and back. Around 25% of patients with celiac disease also have dermatitis herpetiformis (Collin et al., 1997) while 90% of patients with dermatitis herpetiformis have celiac disease (Hill et al., 2000). Adults may also experience depression and a general feeling of illness, while children are frequently irritable and may have delayed growth and development (Green, 2006).
Symptoms of celiac disease include:
In addition, many other autoimmune diseases are associated with celiac disease including type 1 diabetes and autoimmune thyroid disease (Fasano, 1996). Research has also linked mannose-binding lectin deficiency to celiac disease, as the MBL2 genotype is present in a higher percentage of celiac patients than healthy controls. The reason behind the linkage is not fully understood, although the function of MBL in clearing away apoptitic cells and immune complexes, such as antibodies bound to gut epithelial cells, may play a role (Boniotto et al., 2005).
The only way to diagnose celiac disease in the past has been a biopsy of the small intestine to look for villi atrophy. Pancreatic insufficiency, celiac sprue, and Crohn's disease are three most common conditions that cause malabsorption in the United States, so it is important to test specifically for celiac disease before making a diagnosis. The development of less invasive antibody blood tests has made screening for the disease easier, although biopsies are still used to confirm the disease. Celiac disease tests measure the amount of autoantibody in the blood produced in response to gluten and gliadin, and are available in both an IgA and IgG form. These autoantibodies cause inflammation and damage to the intestinal wall. IgA found on the surface of the epithelium and IgG found in body fluid are two of the five classes of antibodies the immune system makes. IgA tests tend to be more specific, but IgG tests are important as a follow-up test to avoid false negatives because celiac disease and IgA deficiency are linked (Mayo Clinic, 2006).
Autoantibody blood tests that are available include (Celiac Disease Tests, 2006):
Anti-tissue Transglutaminase Antibody (tTG), IgA: Tissue transglutaminase is an enzyme responsible for crosslinking certain proteins. It has been identified as the antigen that the body responds to when it creates Anti-EMA antibodies. Gliadin, a peptide derivative of gluten, triggers the development of tTG autoantibodies. Although “tissue” is in the name of this autoantibody, it nevertheless involves testing blood and not tissue.
Anti-Endomysial Antibodies (EMA), IgA: Endomysium is the thin connective tissue layer that covers individual muscle fibers. Anti-Endomysial antibodies are developed in reaction to the ongoing damage to the intestinal lining. Almost 100% of patients with active celiac disease and 70% of patients with dermatitis herpetiformis (another gluten-sensitive enteropathy that causes an itchy burning blistering rash on the skin) will have Anti-EMA, IgA antibodies. Anti-tTG and Anti-EMA antibodies measure the same tissue damage.
Anti-Gliadin Antibodies (AGA), IgG and IgA: Gliadin is part of the gluten protein found in wheat (similar proteins are found in rye, barley, and oats). AGA is an autoantibody against the gliadin portion. It is created by those who are sensitive to it when they are exposed to gluten over a period of time.
Anti-Reticulin Antibodies (ARA), IgA: Anti-ARA is not ordered as frequently because it is not as specific or sensitive as the other autoantibodies. It is found in about 60% of celiac disease patients and about 25% of patients with dermatitis herpetiformis. When used, ARA is ordered along with other celiac disease tests to help diagnose celiac disease.
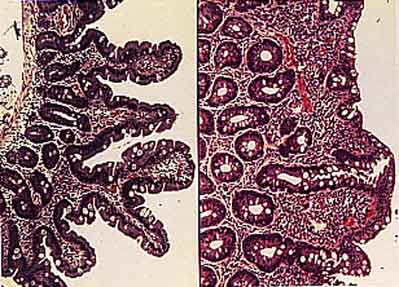
Figure 3 Villi Atrophy (image from Dr. David Elliot, permission pending)
Hematoxylin-eosin stained cross-sections of intestinal villi: normal epithelium (left) and celiac epithelium (right), characterized by infiltration of intraepithelial lymphocytes and flattened villi.
Currently, the only treatment for celiac disease is a gluten-free diet. Gluten is found in wheat, and in smaller amounts in barley, rye, and oat products. Gluten is widely used in commercial and processed foods where wheat flour is used as a thickener in porcessing some meat, vegetable, fruit, and dairy products (Farrell and Kelly, 2002). There are many web resources listed at the bottom of this webpage to aid in managing a gluten-free diet, although direct patient counseling with a registered dietician is recommended. Dietary avoidance of gluten leads to symptom improvement in 70% of patients within 2 weeks (Pink and Creamer, 1967). Titers of EMA, Ttg, and AGA can be used to monitor compliance and response to therapy because they decrease after the initiation of a gluten-free diet. After 3 to 6 months, antibody levels usually become undetectable with strict gluten avoidance (Farrell and Kelly, 2002). However, complete histological resolution of small bowel inflammation may take up to 2 years in some individuals (Greft et al., 1988).
It can be challenging avoiding gluten sometimes, especially when eating out, traveling, or when food labels are unclear. If a patient with celiac disease accidently ingests gluten, an acute celiac crisis can occur manifested by severe diarrhea, dehydration, weight loss, acidosis, hypocalcemia, and hypoproteinemia (Lloyd-Still et al., 1972). Rarely, patients with a high severity of celiac disease could go into gliadin shock. Intravenous corticosteroid therapy is required for critically ill patients. Other immosuppressant agents, including azathioprine and cyclosporin A can also be used (Rolny et al., 1999 and Vaidya et al., 1999).
New methods of treatment aimed at designing orally active drugs are being developed as the pathophysiology of celiac disease becomes better understood. One promising area of current research is in peptidases, including bacterial prolyl endopeptidase, which can degrade gluten into nontoxic components, analogous to the use of lactase in individuals with lactose intolerance. However, a challenge to the enzymatic approach is the timing as the enzyme would need to be in the right place in the intestine at the exact time food is being digested (Shan, 2002). A drug could also act by inhibiting activation of T cells or by binding up tTGase so that gluten was not broken down into toxic peptides. A problem with inhibiting tTGase is that the enzyme plays an important role in the normal repair of the stomach lining, so inhibiting it could reduce the body’s maintenance of that tissue and thus cause intestinal bleeding. Another approach is to develop a drug that acts as analogs of gluten peptides to inhibit the peptides from binding to HLA-DQ2 or DQ8. This is probably the safest possible therapy because other inhibiting these binding sites will not inhibit all antigen presentation (Kim et al., 2004). It is likely that a single therapy might not be able to break the pathological chain of events. The best treatment might be a combination of drugs that weaken several steps, like the triple cocktail approach to treating AIDS.
If the prevalence of celiac disease is 1 in every 133 people, then chances are there are an estimated 13 people among the 1700 students at Davidson College who are affected. However, celiac disease often goes undiagnosed or misdiagnosed because clinical manifestations range from asymptomatic to severe malabsorption. I chose the topic of celiac disease because two of my friends in the Davidson class of 2006 have celiac disease. I interviewed Sabrina Rissing about how she deals with her gluten intolerance and its affect on her college experience. Click on the link below her picture to view her story reprinted with permission.

Figure 4: Sabrina Rissing, English Major from Evans, Georgia, Class of 2006.
Click here to read my Interview with Sabrina Rissing
American Dietetic Association
120 South Riverside Plaza, Suite 2000
Chicago, IL 60606–6995
Phone: 1–800–366–1655 or 1–800–877–1600
Email: hotline@eatright.org
Internet: www.eatright.org
Celiac Disease Foundation
13251 Ventura Boulevard, #1
Studio City, CA 91604
Phone: 818–990–2354
Fax: 818–990–2379
Email: cdf@celiac.org
Internet: www.celiac.org
Celiac Sprue Association/USA Inc.
P.O. Box 31700
Omaha, NE 68131–0700
Phone: 1–877–272–4272 or 402–558–0600
Fax: 402–558–1347
Email: celiacs@csaceliacs.org
Internet: www.csaceliacs.org
Gluten Intolerance Group of North America
15110 10th Avenue, SW., Suite A
Seattle, WA 98166
Phone: 206–246–6652
Fax: 206–246–6531
Email: info@gluten.net
Internet: www.gluten.net
Gluten-Free Living (a bimonthly newsletter)
P.O. Box 105
Hastings-on-Hudson, NY 10706
Phone: 914–969–2018
Email: gfliving@aol.com
National Foundation for Celiac Awareness
124 South Maple Street
Ambler, PA 19002
Phone: 215–325–1306
Email: info@celiacawareness.org
Internet: www.celiacawareness.org
North American Society for Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN)
P.O. Box 6
Flourtown, PA 19031
Phone: 215–233–0808
Email: naspghan@naspghan.org
Internet: www.cdhnf.org www.cdhnf.org
Boniotto, M. et al. (2005) “Evidence of a correlation between mannose-binding lectin and celiac disease: a model for other autoimmune diseases.” Journal of Molecular Medicine. 83(4):308-315.
“Celiac Disease.” Lab Tests Online. < http://labtestsonline.org/understanding/conditions/celiac.html> Accessed April 24, 2006.
Collin, P. et al. (1997) “High incidence and prevalence of adult coeliac disease.” Scandinavian Journal of Gastroenterology. 32:1129-1133.
Fasano A. (1996) "Where have all the American coeliacs gone?" Acta. Pediatriac Suppl 412: 20-24.
Farrell, RJ and Kelly CP. (2002) “Celiac sprue.” New England Journal of Medicine. 348:180-188.
Feighery C, Weir et al. (1998)“Diagnosis of gluten-sensitive enteropathy: is exclusive reliance on histology appropriate?” Eur Journal of Gastroenterol Hepatol. 10: 919-925.
Grefte, JM et al.(1988) “Slow and incomplete histological and functional recovery in adult gluten sensitive enteropathy.” Journal of Clinical Pathology. 41:886-891.
Kim, C. et al. (2004) “Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease.” Proc. Nat. Acad. Sci. 2101: 4175-4179.
Logan RFA. “Epidemiology of coeliac disease.” Coeliac disease. Oxford: Blackwell Scientific, 1992:192-194.
Lloyd-Still JD, et al. (1972) “The use of corticosteroids in celiac crisis.” 81:1074-1081.
Mayo Clinic Staff. "Celiac disease."<www.mayoclinic.com>Accessed April 17, 2006.
NIH Consensus Development Conference on Celiac Disease, National Institutes of Health Consensus Development Conference Statement; June 28-30, 2004. http://consensus.nih.gov/2004/2004CeliacDisease118html.htm. Accessed April 20, 2006.
Not, T. et al. (1998) “Celiac disease risk in the USA: high prevalence of antiendomysium antibodies in healthy blood donors.” Scandinavian Journal of Gastroenterology. 33:494-498.
Papadopoulos, George et al. (2001) “Interpaly between genetics and the environment in the development of celiac disease: perspectives for a healthy life.” The Journal of Clinical Investigation. 108(9): 1261-1266.
Pink, IJ and Creamer B. (1967) “Response to a gluten-free diet of patients with coeliac syndrome.” Lancet. 1:300-304.
Rolny, P et al. (1999) “Role of immunosuppressive thearpy in refractory sprue-like disease.” American Journal of Gastroenterology. 94:219-225.
Shan, Lu et al. (2002) “Structural basis for gluten intolerance in celiac sprue.” Science. 297 (5590): 2275-2279.
Westerberg, Dyanne et al. (2006) “New Strategies for Diagnosis and Management of Celiac Disease.” The New England Journal of Medicine. 106(3):145-151.
Vaidya, A et al. (1999) “Azathioprine in refractory sprue.” American Journal of Gastroenterology. 92:1967-1969.
Return to my Immunology Homepage
send questions, comments to camohr@davidson.edu
Copyright 2006 Carolyn Mohr and Davidson College