(treatment, side effects, & corticosteroid-sparing drugs and alternatives)
This webpage was created in honor of my grandmother, who is currently being treated for Giant Cell Arteritis.
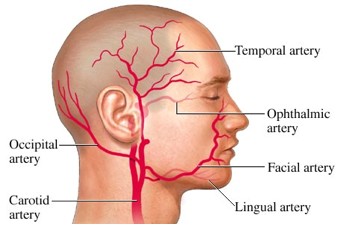
Giant Cell Arteritis (GCA) or temporal arteritis is a chronic inflammatory disorder, also called a necrotizing vasculitis, that affects large and medium-sized arteries that supply the head eyes, and optic nerves (Wagner et al., 2003; Azhar et al., 2005; St Luke’s, 2004). The affected arteries are those that branch from the external carotid artery in the neck or from the aortic arch, particularly the temporal arteries (see Figure 1). In addition to symptoms caused by damage to the cranial arteries, GCA may also have systemic effects (Peng, 2005; Azhar et al., 2005). Untreated, GCA will quickly cause bilateral, permanent vision loss due to ischemic optic neuropathy (the loss of function of all or part of the optic nerve due to disrupted blood flow to the nerve) (Rahman & Rahman, 2005; Wikipedia, 2006 f). Other complications that may accompany GCA include stroke, aortic dissection, myocardial infarctions, and/or aortic aneurysms (Ma-Krupa et al., 2004; Schmidt, 2006). Approximately 50% of GCA patients also have polymyalgia rhematica (PMR), which is characterized by muscle pain and stiffness. However, the connection between the two conditions is unknown (Azhar et al., 2005). GCA is treated with corticosteroids to prevent vision loss and other negative effects; with corticosteroid treatment, GCA does not reduce the life expectancy of affected individuals (Schmidt, 2006).
Figure 1: Arteries commonly affected in GCA. This diagram shows some of the arteries branching from the carotid arteries that are commonly affected in GCA. Notice that other affected arteries may include those originating from the aortic arch and are not shown in this image
Image courtesy of WebMD, 2001 <http://www.webmd.com/hw/health_guide_atoz/zm2207.asp>. Permission pending.
GCA is most common in Caucasian populations, particularly those of Scandinavian or Northern European descent (Rahman & Rahman, 2005). Some people may have a genetic predisposition to GCA; there are often increased numbers of the MHC antigens HLA-DR1, HLA-DR3, HLA-DR4, and HLA-DR5 in GCA. Also, the HLA-DRB1*04 allele is expressed in many GCA patients (Rahman & Rahman, 2005). GCA is primarily seen in women over the age of 50. The incidence is 24.2 per 100,000 women over 50 and 8.2 per 100,000 men over 50 (Azhar et al., 2005). The likelihood of GCA increases will age, with the mean age of presentation at 71 years (Rahman & Rahman, 2005).
Individuals with GCA may present a wide range of clinical symptoms. The most common symptom is recent or atypical headaches, which occur in 60-90% of GCA cases. Other physical symptoms include scalp tenderness near the temporal arteries, temporal artery abnormalities such as reduced or absent pulsation, erythema (redness caused by inflammation), nodularity, or swelling that can be seen upon physical examination, unexplainable fever, jaw claudication (pain in the jaw near the temporo-mandibular joint after chewing for a short time), and eye-related abnormalities such as impaired sight, diplopia (double vision), or a “pale and swollen” optic disk (Azhar et al., 2005; Rahman & Rahman, 2005; Wikipedia, 2006 a & b).
There are five diagnostic criteria for GCA as defined by the American College of Rheumatology. If a patient displays three of these five criteria, the patient is diagnosed with GCA. The criteria are:
1) Age at onset of 50 years or older
2) The onset of new headache
3) Temporal artery abnormality characterized by tenderness or reduced pulsation (see Figure 2)
4) Raised erythrocyte sedimentation rate (ESR) above 50 mm/h by the Westergren method
5) Abnormal temporal artery biopsy (TAB) characterized by mononuclear cell infiltration, granulomatous inflammation, elastic lamina fragmentation, and often the presence of multinucleated giant cells (Rahman & Rahman, 2005; Azhar et al., 2005; Schmidt, 2006) (see Figure 3).
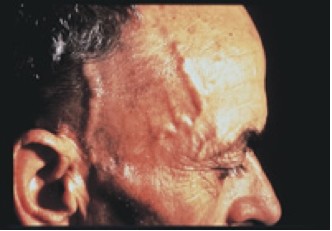
Figure 2: An abnormal temporal artery in a GCA patient. This photograph shows the swelling and thickening of the temporal artery in a GCA patient
Image courtesy of Docken, 2004 <http://www.rheumatology.org/public/factsheets/GCA_new.asp?aud=pat>.
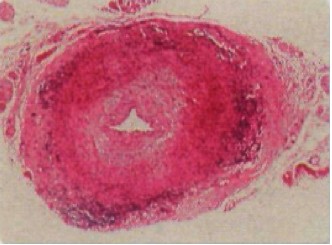
Figure 3: Image of the temporal artery from TAB of a GCA patient. One can see the occlusion of the arterial lumen, fragmentation of the elastic lamina, the “patchy degeneration of smooth muscle,” and the inflammatory infiltrates that are commonly seen in GCA (Azhar et al., 2005).
However, there are problems with the accuracy and applicability of these five diagnostic criteria. First, headache (and scalp tenderness) may be caused by other conditions that are unrelated to GCA (Hayreh & Zimmerman, 2003). Second, physical examination of the temporal artery is imprecise. Patients with and without GCA have shown normal and abnormal temporal artery upon physical examination (Rahman & Rahman, 2005). Third, normal or low ESR does not remove the possibility of GCA because ESR is affected by age, sex, and other factors such as diabetes and infection. Fourth, although TAB is currently considered the best diagnostic tool for GCA, even a TAB may yield a misdiagnosis. For instance, a false-negative result of TAB may occur because of ‘skip areas’ of arteritis in the superficial temporal arteries so that if the entire length of the artery is not checked, the signs of GCA may be missed (Hayreh & Zimmerman, 2003). False negative diagnoses occur for approximately 5-13% of TABs (Rahman & Rahman, 2005). Also, there are other conditions that cause temporal artery abnormalities that may be misdiagnosed as GCA based on the TAB results. Yet, despite the possibility for a false positive diagnosis, a positive TAB is currently considered proof of GCA (Hayreh & Zimmerman, 2003; Rahman & Rahman, 2005). Finally, the use of systemic symptoms in itself is problematic because approximately 21% of GCA patients do not have systemic manifestations of the disease at the time of vision loss, even though they do have an abnormal TAB (Hayreh & Zimmerman, 2003).
In addition to problems caused by the inaccuracy of some of these tests, the five criteria put forward by the American College of Rheumatology are insufficient because they do not include other diagnostic tools that may be more accurate in diagnosing GCA (Rahman & Rahman, 2005). There are a range of diagnostic tests for GCA, none of which are 100% accurate, but can be used in combination to provide an accurate diagnosis of GCA. For instance, ESR and CRP are usually used in combination because although neither is specific for GCA, in conjunction, they can detect GCA with 97% accuracy (Hayreh & Zimmerman, 2003). These diagnostic tests/criteria include:
-TAB (discussed above) is a relatively safe procedure and requires the removal of close to a 1cm section of the temporal artery (Schmidt, 2006). However, there may be some complications due to surgery (Hayreh & Zimmerman, 2003; Rahman & Rahman, 2005).
-Westergren ESR (discussed above) is a non-specific measure of inflammation. Normal ESR for individuals over 50 years is 20mm in men and 30mm in women. The ESR is elevated above 50mm in 90% of GCA patients (Azhar et al., 2005).
-C-reactive protein (CRP), which is a non-specific marker of inflammation, is often elevated in GCA above 2.45mg/dL (Hayreh & Zimmerman, 2003; Azhar et al., 2005). The measure of CRP may be better than ESR because it is sensitive, reproducible, and quantitative, it changes with disease activity more rapidly than ESR, and it is not altered by factors such as age, sex, and others.
-Various imaging techniques including ultrasonography, angiography, MRI, magnetic resonance angiography, positron emission tomography, and CT are now being used, with varying degrees of accuracy and invasiveness (Schmidt, 2006).
-A plasma IL-6 test has recently been suggested as a marker for GCA that may be more sensitive than ESR. IL-6 levels above 6.1pg/ml are indicative of GCA (Azhar et al., 2005).
-A serum platelet count above 400 x 10^3/μl is indicative of thrombocytosis and occurs in approximately 60.4% of CGA cases (Hayreh & Zimmerman, 2003).
-Fluorescein fundus angiography can be used to determine the quality of circulation in the posterior ciliary artery, central retinal artery, retina, and optic nerve (Hayreh & Zimmerman, 2003).
-Other hematologic tests may be used because GCA patients typically have elevated white blood cell counts and below normal levels of hemoglobin and hematocrit (Hayreh & Zimmerman, 2003).
In GCA, granulomas (formed by activated CD4+ T cells and macrophages) and (sometimes) multinucleated giant cells infiltrate the walls of the affected arteries (Weyand et al., 2004; Rahman & Rahman, 2005). Various immune cells produce a range of cytokines that promote inflammation, thus levels of TNF-α, IFN-γ, IL-6, and IL-1B are usually elevated in GCA. Ultimately, the various effector functions of the activated macrophages and T cells cause tissue injury, including damage to the arterial elastic lamina, which leads to luminal (narrowing of the blood vessel), tissue ischemia (the restriction of blood supply, which may cause tissue damage and/or necrosis), and systemic inflammation (see Figure 4) (Weyand & Goronzy, 2003; Ma-Krupa et al., 2005; Rahman & Rahman, 2005; Wikipedia, 2006 e & h).
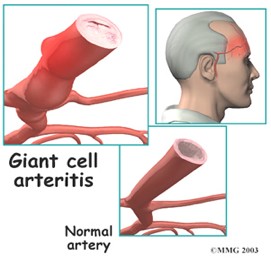
Figure 4: Diagram of a normal artery and an artery affected by GCA. The top left diagram depicts an artery affected by GCA in which there is swelling and occlusion of the vessel. The bottom right diagram shows a normal, unaffected vessel, which does not show signs of inflammation nor of blockage.
Image courtesy of <http://www.allaboutarthritis.com/AllAboutArthritis/layoutTemplates/html/en/contentdisplay/document/condition/arthritis/clinicalArticle/polymyalgia_arteritis.htm>. Permission Pending.
The initial trigger of the chronic inflammatory response seen in GCA is unknown. At one time, scientists believed that infection might trigger GCA (Russo et al., 1995). Since then, studies have shown that bacteria do not have a role in promoting GCA. However, there is still the possibility that “bacterial degradation products” may stimulate the chronic inflammation seen in GCA (Renko et al., 2003). Similarly, it is possible that microbial infection may stimulate the acute-phase response in GCA, but current evidence is inconclusive (Rahman & Rahman, 2005).
The adventitial dendritic cells play a key role in recruiting and activating T cells (the adventitia is the outermost connective tissue covering of arteries) (Ma-Krupa et al., 2005; Wikipedia adventitia). These indigenous dendritic cells secrete cytokines to recruit and locally activate CD4+ T cells against some specific antigen (Ma-Krupa et al., 2005; Rahman & Rahman, 2005). Adventitial and medial phagocytes also release pro-inflammatory proteins (S100 proteins) that further contribute to the arterial inflammation response seen in GCA (Foell et al., 2004). The crucial role of dendritic cells as the antigen presenting cells in the adventitia was shown using human GCA artery-severe combined immunodeficiency (SCID) mouse chimeras. In these GCA SCID mice, GCA was eliminated by depletion of CD83+ dendritic cells. From this evidence, it was proposed that the adventitial dendritic cells initiate and maintain the T cell response seen in GCA arteries and are involved in breaching tissue tolerance the affected area (Ma-Krupa et al., 2004). An additional study supporting the role of dendritic cells in GCA found that adventitial dendritic cells and activated CD4+ cells usually co-localize to the granulomatous infiltrates seen in GCA (Wagner et al., 2003).
These adventitial dendritic cells have a series of Toll-like receptors (TLRs), including TLR-4, that are involved in initiating inflammation of the artery wall (Ma-Krupa et al., 2005). Studies have shown that the indigenous dendritic cells and macrophages in the adventitia expressing TLR-4 also expressed high levels of TNF-α. This finding supports the conclusion that the TLR-4 receptor is involved in the signaling pathways that stimulate TNF-α production, thus contributing to the immune response seen in GCA (Wagner et al., 2003).
The CD4+ T cells that are recruited and activated by adventitial dendritic cells remain in the arterial wall and secrete cytokines, such as interferon (IFN)-γ, which activate macrophages (Weyand & Goronzy, 2003). Some of these cytokines, such as IFN-γ, are also involved in the formation of multinucleated giant cells (Rahman & Rahman, 2005).
The macrophages that are activated by T cells produce additional pro-inflammatory cytokines (Weyand & Goronzy, 2002). In the adventitia, these activated macrophages produce IL-1B and IL-6, which contribute to systemic inflammation and an acute-phase immune response (Rahman & Rahman, 2005; Weyand & Goronzy, 2003). In the media, activated macrophages cause oxidative stress, which causes smooth muscle cell apoptosis, nitration of endothelial cells, and damage to the matrix (Weyand & Goronzy, 2002; Weyand et al., 2004). Activated macrophages also produce growth factors that cause intimal hyperplasia (increased production of cells in the intima, or inner layer of the artery) (Rahman & Rahman, 2005; Wikipedia, 2006 c & d). Intimal hyperplasia may also involve giant cells, particularly in GCA patients with higher levels of IFN-γ (Rahman & Rahman, 2005). Intimal hyperplasia causes occlusion (closing) of the arterial lumen, thus contributing to the ischemia seen in GCA (Weyand & Goronzy, 2002; Weyand et al., 2004; Wikipedia, 2006 g).
Tissue damage caused by activated macrophages is primarily seen in the elastic lamina of the arterial walls, whereas little tissue damage is seen in the adventitia (Rahman & Rahman, 2005). Activated macrophages in the media-intima junction produce matrix metalloproteinases (MMP), which digests the elastic lamina. It has been suggested that the fragmented elastin may serve as an autoimmune target in GCA, but this has not been proven (Gillot et al., 1997). Yet, it is clear that fragmentation of the elastic lamina is an important factor in GCA because arteries with little elastic lamina, such as the intracranial arteries, are usually not affected in GCA (Rahman & Rahman, 2005).
Varying levels of cytokines and the intensity of the immune response may contribute to the severity of GCA. For instance, GCA patients with stronger systemic inflammation have higher levels of the pro-inflammatory cytokines that are normally involved in GCA such as IL-1B, IL-6, and TNF-α (Hernandez-Rodriguez et al., 2004). Also, patients with higher levels of IFN-γ than are seen in average GCA cases typically have more ischemic symptoms (Rahman & Rahman, 2005). Similarly, the intensity of induced adhesion molecules VCAM-1 and E-selectin was positively correlated with the intensity of the inflammation in GCA patients (Cid et al., 2000).The role of CD8+ T cell in GCA is unclear. Some studies suggested that increased CD8+ T cell numbers were involved in the pathogenesis of GCA (Martinez-Taboada et al., 1996 b), whereas other studies suggested that there were decreased numbers of CD8+ T cells in GCA (Martinez-Taboada et al., 2001; Salvarani et al., 1995). A more recent study of a small number of GCA patients found that CD8+ T cell numbers were not altered in GCA (Martinez-Taboada et al., 2001). Clearly, future studies will need to be conducted to determine the role, if any, of CD8+ T cells in GCA.
Although the immune response in GCA excludes B cells, some evidence suggests that a humoral immune response may play a role in the pathogenesis of GCA. Specifically, immunoglobulin and complement deposition is often found in the wall of the affected arteries. However, studies have not determined the definite presence of antibodies against any specific molecule/protein present in the inflamed arteries. It is possible that the disposition of complement and antibodies is non-specific and results from increased endothelial permeability that occurs in affected arteries in GCA (Martinez-Taboada et al., 1996 a; Rahman & Rahman, 2005).
Corticosteroid treatment is typically started as soon as GCA is suspected, even before the diagnosis is confirmed with diagnostic tests such as TAB (Schmidt, 2006). After steroid therapy is begun, a window of approximately eight days remains during which time inflammation and other histological indicators of GCA can be seen in TAB in order to confirm the diagnosis (Azhar et al., 2005). The aim of corticosteroid treatment is to suppress the inflammatory response and to minimize the ischemic complications seen in GCA (Rahman & Rahman, 2005).
The most commonly prescribed corticosteroid for GCA is prednisone, a synthetic analogue of cortisol. Corticosteroids exert their effects by entering the cytoplasm of cells where they bind to a steroid receptor and displace a regulatory protein. Then, the corticosteroid:receptor complex enters the nucleus and activates transcription of up to 1% of the genes in the genome (the expression of a small number of genes will simultaneously be decreased). The altered regulation of a variety of genes produces the many effects of corticosteroid treatment (Janeway et al., 2005).
Interestingly, despite the fact that corticosteroids are the standard treatment for GCA, no placebo-controlled studies of the drug’s impact on GCA have ever been conducted, and are now unethical because of the serious consequences of untreated GCA (Schmidt, 2006). However, various studies have begun to elucidate some of the ways corticosteroids are able to limit the effects of GCA as well as ways that corticosteroids are ineffective at treating this disease.In treating GCA, corticosteroids are able to prevent blindness in GCA patients for whom treatment is started immediately, but will not reverse vision loss caused by untreated GCA (Hayreh & Zimmerman, 2003; Piptone et al., 2005). Corticosteroids can also suppress much of the inflammation response and improve endothelial function (Weyand & Goronzy, 2003; Gonzalez-Juanatey et al., 2006). But, despite the generally positive effect of corticosteroid therapy, there are limitations to this treatment for GCA. For instance, although corticosteroids are able to suppress the systemic inflammation seen in GCA, they do not eradicate the immune responses that occur in arterial walls (Weyand & Goronzy, 2003). This occurs for multiple reasons. First, corticosteroid treatment lowers IL-6 and IL-1B production, but it does not correct the underlying reason for the production of these cytokines (Roche et al., 1993). Second, treatment only slightly decreases the production of IFN- γ and does not completely block the effects of activated macrophages (Weyand et al., 2002; Brack et al., 1997). Finally, although corticosteroid treatment has been shown to reduce the expansion of the inducible adhesion molecules VCAM-1 and E-selectin, it does not eliminate the production of these molecules completely (Cid et al., 2000).
Partially because corticosteroids have not been studied in the context of GCA, there are no officially recommended treatment regimes or guidelines for corticosteroid treatment of GCA. Instead, the dosage of steroids depends on the severity of symptoms and test results of the individual patient. Typically, the goal in treatment is to progressively decrease the dosage of steroids while effectively suppressing the disease. However, as steroid therapy is reduced or terminated, GCA may reappear. ESR and CRP measures are able to detect this re-emergence, but there are often no systemic symptoms that reappear with the condition (Hayreh & Zimmerman, 2003). Thus, GCA patients must be continually monitored using ESR and CRP during dosage reduction and after treatment is completed.
A general treatment regime for GCA patients that many physicians follow begins with a 70mg/day prednisone dose. After the first week, the dose is reduced by 10mg every week until the dosage reaches 20mg/day (Schmidt, 2006). However, some physicians recommend that the high initial dose be maintained for two weeks (Rahman & Rahman, 2005). Once the 20mg/day level is reached, the dosage is lowered by 2.5mg per week. Then, when the dosage reaches 10mg/day, it is reduced by 1mg every month depending on the symptoms and test results (Schmidt, 2006). Most patients remain on corticosteroids for 1-2 years, although some may require long-term steroid treatment (Schmidt, 2006; Piptone et al., 2005). The reason that treatment of GCA is usually short in duration is because GCA is a self-limiting disease for most patients (although the mechanism for this limitation is unknown) (Azhar et al., 2005; Schmidt, 2006). Longer corticosteroid treatment is typically required for patients with more severe GCA; these patients usually have cytokine levels (such as TNF-α) above most GCA patients (Hernandez-Rodriguez et al., 2004).
Side Effects
Despite the success of corticosteroid treatment for GCA, there are many negative side effects that are associated with the drug; these negative effects are particularly prevalent in elderly GCA patients (Azhar et al., 2005).
Corticosteroid side effects may include osteoporosis, gastrointestinal disturbances (dyspepsia, peptic ulcers, etc.), glucose intolerance or diabetes mellitus, arterial hypertension (high blood pressure), increased homocysteine concentrations, muscle wasting, weight gain, increased skin vulnerability caused by thinning of the skin, various mental disturbances (depression, psychosis, or euphoria), infection, glaucoma, cataracts, and others (Rahman & Rahman, 2005; Schmidt, 2006; Janeway et al., 2005; Martinez-Tobaoda et al., 2003).
When a GCA patient is on corticosteroid treatment, drug side effects should be monitored and some may require treatment. Suggested tests to monitor steroid side effects include measurements of bone mineral density and regular monitoring of blood pressure and glucose levels (Schmidt, 2006).
There are a range of lifestyle and drug treatments to address the side effects of corticosteroids. To prevent osteoporosis, GCA patients undergoing steroid treatment should take 1200-1500 mg of calcium and 800 IU of vitamin D per day and, if necesary, bisphosphonates or other hormones may be prescribed (Azhar et al., 2005; Schmidt, 2006; Rahman & Rahman, 2005; Turbin & Kupersmith, 1999). Unfortunately, bisphosphonates have additional side effects including gastrointestinal problems (indigestion, diarrhea or constipation, and abdominal pain), musculoskeletal pain, and headache (Rahman & Rahman, 2005). In addition to medical treatments to prevent osteoporosis, it is recommended that GCA patients should stop smoking (which will also reduce the risk of vascular complications), decrease alcohol consumption, and begin some form of weight-bearing exercise (Schmidt, 2006). In order to prevent gastric problems, patients may receive histamine (H(2))-blocking agents or protein-pump inhibitors (Rahman & Rahman, 2005). Also, insulin and antihypertensive drugs may be prescribed if necessary (Turbin & Kupersmith, 1999). Additionally, GCA patients may be given folic acid and/or vitamin B12 to reduce homocysteine concentrations, which are elevated in GCA and are further increased when a patient begins corticosteroid treatment (Martinez-Tobaoda et al., 2003).In addition to steroid treatment, low doses of aspirin (an anti-platelet aggregating agent) is thought to minimize the risk of vision loss and other ischemic complications because thrombocytosis is usually seen in GCA (Hayreh & Zimmerman, 2003; Rahman & Rahman, 2005; Piptone et al., 2005). Although the benefits of aspirin have not been proven beyond 3 months of treatment, physicians expect that the benefits will continue. Unfortunately, if treatment involves corticosteroids in combination with aspirin, a protein pump inhibitor should also be taken to prevent gastric ulcers (Schmidt, 2006).
Corticosteroid-Sparing Drugs and Alternatives
Because of the negative side effects associated with corticosteroid treatment of GCA, there has been a largely unsuccessful and continuing search for corticosteroid-sparing drugs and alternatives.
For instance, methotrexate treatment was proposed as a corticosteroid-sparing drug, but conflicting results as to its effectiveness in this role have been obtained (Schmidt, 2006). In 2001, two randomized, controlled, double-blind trials were conducted; one found that methotrexate did not have a corticosteroid-sparing effect (Spiera et al.), whereas the other found that combined treatment with methotrexate and corticosteroid was more effective than corticosteroid treatment alone (Jover et al.). Unfortunately, methotrexate, like corticosteroids, has a range of potential side effects including nausea, liver toxicity, ulcerative stomatitis (inflammation of the mouth), and pneumonitis (Schmidt, 2006; Wikipedia, 2006 i). The effects of methotrexate in GCA treatment are still being studied (Piptone et al., 2005). However, despite the possibility that methotrexate is ineffective or only minimally effective at treating GCA, the drug is often used in combination with corticosteroids if the steroid treatment must be maintained at levels of 10mg/day or higher for an extended period of time (Schmidt, 2006). Regrettably, no other immunosuppressive drugs have been conclusively shown to have a corticosteroid-sparing effect to this date (Schmidt, 2006). Some of the steroid-sparing drugs that were considered included azathioprine, cyclosporin, and dapsone (Rahman & Rahman, 2005).
Currently, nonsteroidal anti-inflammatory drugs (NSAIDs) are sometimes used in combination with corticosteroid treatment or alone in cases of mild GCA (Schmidt, 2006). Also, other alternatives to corticosteroids are being explored. For example, at one time infliximab (an anti-TNF-α antibody) was considered as an appropriate treatment, particularly in severe or steroid-resistant cases, but it is now considered unhelpful for the treatment of GCA (Rahman & Rahman, 2005; Piptone et al., 2005; Schmidt, 2006). Fortunately other promising drugs are being studied. For instance, Weyand et al. (2002) found that acetylsalicylic acid, like corticosteroids, suppressed the production of pro-inflammatory cytokines, particularly IFN- γ in GCA, thus might be used as an alternative or additional treatment (Schmidt, 2006).
Azhar SS, Tang RA, Dortheo EU. 2005. Giant cell arteritis; Diagnosing and treating inflammatory disease in older adults 60(8):26-30.
Brack A, Rittner HL, Younge BR, Kaltschmidt C, Weyand CM, Goronzy JJ. 1997.
Glucocorticoid-mediated repression of cytokine gene transcription in human arteritis-SCID chimeras [abstract]. In The Journal of Clinical Investigation 99(12):2842-50. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
Cid MC, Cebrian M, Font C, Coll-Vinent B, Hernandez-Rodriguez J, Esparza J, Urbano-Marquez A, Grau JM. 2000. Cell adhesion molecules in the development of inflammatory infiltrates in giant cell arteritis: inflammation-induced angiogenesis as the preferential site of leukocyte-endothelial cell interactions [abstract]. In Arthritis Rheumatology 43(1):184-94. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=10643715&query_hl=2&itool=pubmed_docsum>. Accessed 2006 April 10.
Docken W. 2004. Giant Cell Arteritis. American College of Rheumatology. <http://www.rheumatology.org/public/factsheets/GCA_new.asp?aud=pat> Accessed 2006 April 14.
Foell D, Hernandez-Rodriguez J, Sanchez M, Vogl T, Cid MC, Roth J. 2004. Early recruitment of phagocytes contributes to the vascular inflammation of giant cell arteritis [abstract]. In Journal of Pathology 204(3):311-6. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
Gillot JM, Masy E, Davril M, Hachulla E, Hatron PY, Devulder B, Dessaint JP. 1997. Elastase derived elastin peptides: putative autoimmune targets in giant cell arteritis [abstract]. In Journal of Rheumatology 24(4):677-82. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
Gonzalez-Juanatey C, Llorca J, Garcia-Porrua C, Sanchez-Andrade A, Martin J, Gonzalez-Gay MA. 2006. Steroid therapy improves endothelial function in patients with biopsy-proven giant cell arteritis [abstract]. In The Journal of Rheumatology 33(1):74-8. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=16395753&query_hl=6&itool=pubmed_docsum>. Accessed 2006 April 10.
Hayreh SS & Zimmerman B. 2003. Management of giant cell arteritis. Our 27-year clinical study: new light on old controversies. Ophthalmologica 217(4): 239-259.
Hernandez-Rodriguez J, Segarra M, Vilardell C, Sanchez M, Garcia-Martinez A, Esteban MJ, Queralt C, Grau JM, Urbano-Marquez A, Palacin A, Colomer D, Cid MC. 2004. Tissue production of pro-inflammatory cytokines (IL-1beta, TNFalpha and IL-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis [abstract]. In Rheumatology (Oxford) 43(3):294-301. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14679293&query_hl=9&itool=pubmed_docsum>. Accessed 2006 April 10.
Janeway CA, Travers P, Walport M, Shlomchik, MJ. 2005. ImmunoBiology, Sixth Edition. New York: Garland Science Taylor and Francis Group.
Jover JA, Hernandez-Garcia C, Morado IC, Vargas E, Banares A, Fernandez-Gutierrez B. 2001. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial [abstract]. In Annals of Internal Medicine 134(2):106-14. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=11177313&query_hl=11&itool=pubmed_docsum>. Accessed 2006 April 10.
Ma-Krupa W, Jeon MS, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. 2004. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis [abstract]. In The Journal of Experimental Medicine 199(2): 173-83. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
Ma-Krupa W, Kwan M, Goronzy JJ, Weyand CM. 2005. Toll-like receptors in giant cell arteritis [abstract]. In Clinical Immunology 115(1):38-46. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
a. Martinez-Taboada V, Brack A, Hunder, GG, Goronzy JJ, Weyand CM. 1996. The inflammatory infiltrate in giant cell arteritis selects against B lymphocytes [abstract]. In Journal of Rheumatology 23(6):1011-1014. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=8782132&query_hl=17&itool=pubmed_docsum>. Accessed 2006 April 10.
b. Martinez-Taboada VM, Goronzy JJ, Weyand CM. 1996. Clonally expanded CD8 T cells in patients with polymyalgia rheumatica and giant cell arteritis [abstract]. In Clinical Immunology and Immunopathology 79(3):263-70. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=8635285&query_hl=19&itool=pubmed_docsum>. Accessed 2006 April 10.
Martinez-Taboada VM, Blanco R, Fito C, Pacheco MJ, Delgado-Rodriguez M, Rodriguez-Valverde V. 2001. Circulating CD8+ T cells in polymyalgia rheumatica and giant cell arteritis: a review [abstract]. In Seminars in Arthritis and Rheumatism 30(4):257-71. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
Martinez-Taboada VM, Bartolome MJ, Fernandez-Gonzalez MD, Blanco R, Rodriguez-Valverde V, Lopez-Hoyos M. 2003. Homocysteine levels in polymyalgia rheumatica and giant cell arteritis: influence of corticosteroid therapy [abstract]. In Rheumatology (Oxford). 42(9):1055-61. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
Peng S. 2005. Temporal arteritis. Medline Plus. <http://www.nlm.nih.gov/medlineplus/ency/article/000448.htm> Accessed 2006 April 14.
Piptone N, Boiardi L, Salvarani C. 2005. Are steroids alone sufficient for the treatment of giant cell arteritis? [abstract]. In Best Practice & Research; Clinical Rheumatology. 19(2):277-92. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=15857796&query_hl=24&itool=pubmed_docsum>. Accessed 2006 April 10.
Rahman W & Rahman FZ. 2005. Giant cell (temporal) arteritis: an overview and update. Survey of Ophthalmology 50(5): 415-28.
Renko J, Kalela A, Karhunen PJ, Helin H, Sillanaukee P, Nikkari S, Nikkari ST. 2003. Do temporal arteritis lesions contain bacterial DNA? [abstract]. In European Journal of Clinical Investigation 33(8):657-61. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. 1993. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis [abstract]. In Arthritis Rheumatology 36(9):1286-94. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed >. Accessed 2006 April 10.
Russo MG, Waxman J, Abdoh AA, Serebro LH. 1995. Correlation between infection and the onset of the giant cell (temporal) arteritis syndrome. A trigger mechanism? [abstract]. In Arthritis and Rheumatism 38(3):374-80. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
Salvarani C, Boiardi L, Macchioni P, Rossi F, Tartoni P, Casadei Maldini M, Mancini R, Beltrandi E, Portioli I. 1995. Role of peripheral CD8 lymphocytes and soluble IL-2 receptor in predicting the duration of corticosteroid treatment in polymyalgia rheumatica and giant cell arteritis [abstract]. In Annals of the Rheumatic Diseases 54(8):640-4. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
Schmidt WA. 2006. Current diagnosis and treatment of temporal arteritis. Current Treatment Options in Cardiovascular Medicine 8(2):145-51.
Spiera RF, Mitnick HJ, Kupersmith M, Richmond M, Spiera H, Peterson MG, Paget SA. 2001. A prospective, double-blind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA) [abstract]. In Clinical and Experimental Rheumatology 19(5):495-501. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
St. Luke’s Cataract & Laser Institute. 2004. Temporal arteritis (giant cell arteritis). <http://www.stlukeseye.com/Conditions/TemporalArteritis.asp> Accessed 2006 April 15.
Turbin RE & Kupersmith MJ. 1999. Giant Cell Arteritis [abstract]. In Current Treatment Options in Neurology 1(1):49-56. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=11096695&query_hl=31&itool=pubmed_docsum>. Accessed 2006 April 10.
Wagner AD, Wittkop U, Prahst A, Schmidt WA, Gromnica-Ihle E, Vorpahl K, Hudson AP, Zeidler H. 2003. Dendritic cells co-localize with activated CD4+ T cells in giant cell arteritis [abstract]. In Clinical and Experimental Rheumatology 21(2): 185-92. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
WebMD. 2001. Arteries commonly affected by giant cell arteritis. <http://www.webmd.com/hw/health_guide_atoz/zm2207.asp>. Accessed 2006 April 25.
Weyand CM & Goronzy JJ. 2002. Pathogenic mechanisms in giant cell arteritis [abstract]. In Cleveland Clinic Journal of Medicine 69 Suppl 2:SII28-32. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=12086261&query_hl=34&itool=pubmed_DocSum>. Accessed 2006 April 10.
Weyand CM & Goronzy JJ. 2003. Giant-cell arteritis and polymyalgia rheumatica [abstract]. In Annals of Internal Medicine 139(6):505-15. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=13679329&query_hl=36&itool=pubmed_docsum>. Accessed 2006 April 10.
Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. 2002. Therapeutic effects of acetylsalicylic acid in giant cell arteritis [abstract]. In Arthritis and Rheumatism 46(2):457-66. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=pubmed>. Accessed 2006 April 10.
Weyand CM, Ma-Krupa W, Goronzy JJ. 2004. Immunopathways in giant cell arteritis and polymyalgia rheumatica [abstract]. In Autoimmunity Reviews 3(1):46-53. PubMed <http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=pubmed&dopt=Abstract&list_uids=14871649&query_hl=39&itool=pubmed_docsum>. Accessed 2006 April 10.
Wikipedia. 2006. Wikamedia Foundation.
a. Diplopia <http://en.wikipedia.org/wiki/Diplopia>. Accessed 2006 April 25.
b. Erythema <http://en.wikipedia.org/wiki/Erythema>. Accessed 2006 April 25.
c. Hyperplasia <http://en.wikipedia.org/wiki/Hyperplasia>. Accessed 2006 April 22.
d. Intima <http://en.wikipedia.org/wiki/Intima>. Accessed 2006 April 22.
e. Ischemia <http://en.wikipedia.org/wiki/Ischemia>. Accessed 2006 April 22.
f. Ischemic optic neuropathy <http://en.wikipedia.org/wiki/Ischemic_optic_neuropathy>. Accessed 2006 April 25.
g. Occlusion <http://en.wikipedia.org/wiki/Occlusion>. Accessed 2006 April 22.
h. Stenosis <http://en.wikipedia.org/wiki/Stenosis>. Accessed 2006 April 22.
i. Stomatitis <http://en.wikipedia.org/wiki/Stomatitis>. Accessed 2006 April 25.
Return to Immunology Main Page
Go to Davidson College Main Page
E-mail questions & comments to: jaschwartz@davidson.edu