*This website was produced as an assignment for an undergraduate course at Davidson College.*
Note: This web page should NOT be used for medical advice. Please consult your physician for information on the disease
Interferon-gamma Receptor Deficiency
(IFNgR deficiency)
1. Introduction
Infections with intracellular bacteria such as mycobacteria remain an important
cause of human morbidity and mortality worldwide. Immunologic protection against
such organisms depends on cell mediated immunity, the major effector of which
is the IFNg-activated macrophage. IFNg activates transcription of a large number
of genes that play roles in antiviral activity, apoptosis, antigen processing,
MHC protein expression, and type 1 T helper cell (TH1) development. IFNg also
activates macrophages to kill or restrict growth of microbial targets; thus
the appropriate function of IFNg receptor appears to be important in host defense
against mycobacteria (Rosen et al., 2004).
1.1. IFNg and the IFNg receptor
IFNg is produced predominantly by T cells and NK cells in response
to a variety of inflammatory or immune stimuli, and in general, it stimulates
the development and function of immune effector cells (Janeway et al.,
2005) (for more information on IFNg, you can access
my IFNg protein webpage). IFNg receptors are expressed on almost all
nucleated cells, and show species specificity in their ability to bind IFNg
(Farrar et al., 1993). The functional IFNg receptor is composed of two 90 kDa
IFNgR1 proteins and two 62 kDa IFNgR2 proteins . The human
IFNgR1 gene contains seven exons, and is located on chromosome 6. The extracellular
portion of IFNgR1 contains the IFNg ligand-binding domain; the intracellular
portion contains domains necessary for signal transduction and receptor recycling.
The IFNgR2 gene also contains seven exons, and is located on human chromosome
21. The intracellular IFNgR2 domain is necessary for signal transduction. The
extracellular domain of IFNgR2 interacts with the IFNgR1/IFNg complex, but does
not itself play a major role in ligand binding (Bach et al., 1997)
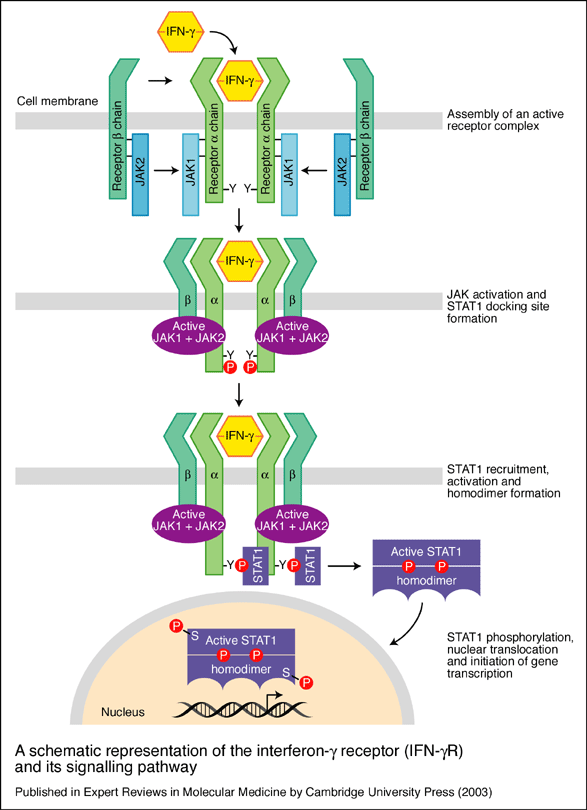
(see Fig. 1) .
Figure 1. A schematic representation of the interferon-g receptor (IFN-gR) and its signalling pathway. The receptor for IFN-g has two subunits: IFN-gR1, the ligand-binding chain (also known as the a chain) and IFN-gR2, the signal-transducing chain (also known as the b chain or accessory factor 1). These proteins are encoded by separate genes (IFNGR1 and IFNGR2, respectively) that are located on different chromosomes. As the ligand-binding (or a) chains interact with IFN-g they dimerise and become associated with two signal-transducing (or b) chains. Receptor assembly leads to activation of the Janus kinases JAK1 and JAK2 and phosphorylation of a tyrosine residue on the intracellular domain of IFN-gR1. This leads to the recruitment and phosphorylation of STAT1 (for ‘signal transducers and activators of transcription’), which forms homodimers and translocates to the nucleus to activate a wide range of IFN-g-responsive genes. After signalling, the ligand-binding chains are internalised and dissociate. The chains are then recycled to the cell surface (Image permit pending).
2. IFNgR deficiency
2.1 WT phenotype vs. IFNgR deficient:
Individuals with defective IFNg receptor expression or function have a widespread defect in macrophage activation, which results in reduced production of TNFa and other proinflammatory cytokines in response to IFNg and endotoxin, defective MHC class II expression in response to IFNg or antigenic stimulation, and reduced ability to present antigen to T cells (Dorman et al., 1998). This importance of IFNg pathways in host defense has been demonstrated in mice with targeted disruptions of the IFNg, IFNGR1, or IFNGR2 genes. IFNgR knockout mice have increased susceptibility to experimental challenge with infectious agents. In contrast to wild-type (WT) mice, IFNg and IFNgR knockout mice develop neither mature granulomas nor protective immunity after experimental infection with mycobacteria (Jouanguy et al., 1999). Thus, IFNgR deficiency is an inherited disorder associated with complications from infections caused by mycobateria, and other microorganisms such as Listeria and Salmonella species.
2.2 Symptoms and Diagnosis
The attenuated strain of Mycobacterium bovis bacille Calmette–Guérin
(BCG) is the most widely used standard vaccine in the world. In most children,
inoculation of live BCG vaccine is harmless. In rare cases, however, vaccination
causes disseminated BCG infection, which may be lethal. Therefore one of the
first symptoms is that affected children invariably develop disseminated (BCG)
infection shortly after inoculation with live BCG vaccine (Roesler et al.,
1999). Later on, the IFNgR deficiency patient will develop a history of severe
or repeated mycobacterial infections that may involve the lungs, lymph nodes,
blood and bone marrow. People with complete IFNGR deficiency have more serious
infections than those with partial IFNGR deficiency. The disease occurs early
in infancy in those with complete IFNGR deficiency. Those with partial deficiency
are more likely to develop illness later in childhood (Fieschi et al.,
2001). Sophisticated laboratory tests measure the amount of interferon gamma
in the blood and show the patient's white blood cells respond poorly, or not
at all, to interferon gamma. Depending on whether the patient has complete or
partial IFNGR deficiency, the blood will have either very high or very low levels
of interferon gamma. Genetic testing can determine whether the patient has mutations
that cause IFNgR deficiency (Roesler et al., 1999).
2.3 Mutations that give rise to IFNgR deficiency
The existence of a genetic component to human mycobacterial disease susceptibility
had long been postulated. Now we know that a variety of IFNgR mutations are
associated with complete or partial IFNgR deficiency. They include nonsense
and splice mutations and frameshift insertions and deletions. All result in
a premature stop codon upstream from the segment encoding the transmembrane
and the extracellular ligand-binding domain, either precluding cell surface
expression of the receptors at the cell surface or by disrupting the IFNg binding
site without affecting surface expression respectively. Phenotype-to-genotype
correlations are being established as more affected individuals are identified.
However, for IFNgR defficiency, the phenotype appears to depend less on which
gene (IFNgR1 vs IFNgR2) is mutated, but rather on the extent to which the mutation
reduces IFNg responsiveness (Dorman et al., 2004).
2.3.1 Complete IFNgR deficiency
Complete absence of IFNg responsiveness is associated with a more severe clinical phenotype. Such affected individuals characteristically have severe disseminated mycobacterial infections that may involve lungs, viscera, lymph nodes, blood, and bone marrow. Onset of first environmentally acquired mycobacterial infection is usually during infancy. Infections are typically caused either by NTM species that are poorly pathogenic in immunocompetent hosts and presumably acquired from environmental exposure, or by BCG acquired by vaccination. In these children, such infections are usually fatal if untreated (Jouanguy et al., 2000).
2.3.2 Partial IFNgR deficiency
Partial deficiency has been shown conclusively to result from one group of
IFNGR1 mutations. These mutations confer partial, but not complete, loss of
IFNg responsiveness, where mutant proteins are expressed on the cell surface
and bind IFNg ligand, but cannot transduce signal due to absence of JAK1 and
STAT1 binding sites. Moreover, the absence of the IFNgR1 recycling motif results
in an increased number of mutant proteins expressed on the cell surface. Residual
IFNg responsiveness is mediated by the normal IFNgR1 proteins expressed from
the normal allele in these individuals. The clinical phenotype associated with
this group is milder than that seen in children with complete absence of IFNg
responsiveness. Environmental mycobacterial infections may first occur during
childhood rather than infancy and may be localized rather than disseminated
(Jouanguy et al., 1997).
2.4 Treatment
Aggressive and prolonged antibiotic therapy can lead to the control of infection in some patients with complete IFNgR deficiency. However, the overall prognosis for these patients is poor since antibiotic therapy apparently does not give sustained remission, and there is continued susceptibility to new mycobacterial infection. IFNg administration would not be expected to be of therapeutic benefit in patients with complete absence of IFNg responsiveness due o the absence of cell surface expression of receptors. Bone marrow transplantation is the only curative treatment available. Scientists are developing methods to add a corrective gene to bone marrow cells that will become granulocytes. They are also working to improve the multi-drug treatment that is the mainstay for IFNgR-deficient patients. Patients with complete IFNgR deficiency may especially benefit from treatment that includes cytokines such as IL-2, IL-12, and GM-CSF. Patients with partial INFgR deficiency have a milder disease, and are usually responsive to appropriate antimicrobial therapy and IFNg administration (Reuter et al., 2002)
3. Conclusion
IFNgR deficiency is listed as a "rare disease" by the Office of Rare
Diseases (ORD) of the National
Institutes of Health (NIH). This means that Interferon gamma receptor deficiency,
affects roughly 200,000 people in the US population. However, the diversity
of the genes and pathogenic mutations involved renders molecular diagnosis challenging.
An accurate and rapid molecular diagnosis is essential for the rational and
efficient treatment of the patient. Indeed, children with complete IFNgR deficiency
do not achieve sustained remission with antibiotics alone and do not respond
to exogenous IFNg, resulting from a lack of functional receptors. The lack of
a simple method for rapidly discriminating between patients with complete or
partial IFNgR deficiency and patients with other genetic etiologies greatly
compromises the management of these patients. Recognition of the role of IFNgR
in human host defense against intracellular pathogens emphasizes the importance
of research to understand the mechanisms by which IFNg activates macrophage
killing of intracellular organisms. Better understanding these mechanisms will
lead to the development of rational preventive and therapeutic strategies
References:
Davidson College Biology Department Home Page
© Copyright 2005, Department of Biology, Davidson College,
Davidson, NC 28036
Send comments, questions, and suggestions to: Daniela
V. Alvarez (davillarrealalvarez@davidson.edu).