Introduction:
MIF (Macrophage Migration Inhibitory Factor) was the first cytokine to be identified (1966), and was originally defined as a compound responsible for “the inhibition of random macrophage migration during the delayed-type hypersensitivity response” (Bloom, 1966). Currently, MIF is known to be a proinflammatory cytokine involved in macrophage and t-cell activation, IgE synthesis, insulin release, carbohydrate metabolism, cell growth and apoptosis, and tumor angiogenesis (Senter et. al, 2002). Its role as an inflammatory mediator has been implicated in several diseases, among them, septic shock, glomerulonephritis, and arthritis. Recent investigation into its role in delivering a macrophage survival signal has implicated MIF as potentially involved in cancer development.
Structure and Receptor Interactions:
MIF is a very unique protein showing “no homology with any other pro-inflammatory cytokine” (Donn and Ray, 2004). Only a single MIF gene exists in humans, located on chromosome 22 (22q11.2). The gene has 3 exons separated by small introns covering, all in all, 1 kB (OMIM 2006). The human MIF cDNA was not cloned until 1989. Despite being the first cytokine discovered, it took a long time to determine MIF’s structure, as it was difficult to isolate enough of the compound from secretions. It is now known that MIF, as visible in figure 1, is a monomer consisting of 115 amino acids, forming a homotrimer. The protein has an alpha/beta structure with six alpha-helices, and three beta-sheets, forming a circular protein with an interior “solvent accessible channel”. This interior hydrophobic core is stabilized by basic residues. MIF undergoes no significant post-translational modification in vivo. MIF’s closest similarity to any known protein is between MIF’s alpha helices and the peptide binding domains of the major histocompatibility complex. However, these domains have not been shown to bear any functional similarity (Bucala, 1996). Current research trying to determine the active site of MIF has shown test chains of amino acids 50-65 to have “MIF-like biological function,” suggesting that this exposed region of the protein is responsible for much of MIF’s activity (Nguyen et. al, 2003).
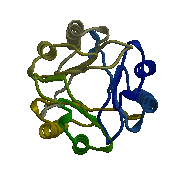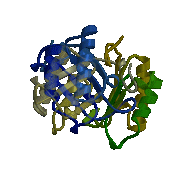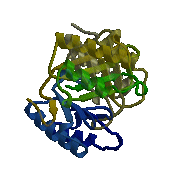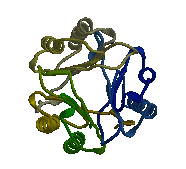
Figure 1: MIF Homotrimer
Source: with permission from Elias Lolis' Lab at Yale
“Until recently [MIF] lacked a recognized transmembrane receptor” (Donn and Ray, 2004). The protein was presumed to require a “receptor-based” mechanism of action as it had been demonstrated to interact with neutrophils, macrophages, and t-cells, providing both activation and survival signals. Recent research into MIF’s protein receptor interactions has pointed to numerous receptor sites. MIF inhibits transcriptional activity of the JAB1 (Jun Activation domain-binding protein) induced activator protein (AP1) (Kleeman et. al, 2000). MIF has also been shown to interact with high affinity with CD74, a transmembrane protein. CD74 initiates a MAP kinase cascade, cell proliferation, and PGE2 production, which eventually promotes monocyte/macrophage activation (Leng et. al, 2003). Lastly, MIF has been demonstrated to interact with p53 gene activators (Fingerle-Rowson et. al, 2003) as well as with TLR-4 gene activators (Roger et. al, 2003).
In addition to its standard receptor based interactions, MIF has also been shown to act as a tautomerase, with an active N-terminal proline. Tautomerase activity can be measured by, for example, tautomerization of dopachrome methyl ester. Interestingly, this process is inhibited by a metabolite of acetaminophen, N-acetyl-p-benzoquinone, as would be expected of a drug for a proinflammatory mediator. This potential role of MIF as an enzyme, in addition to being a cytokine, offers promise for potential therapeutic inhibitory drug development for some of the diseases discussed below (Senter et. al, 2002). This is currently a popular research area for drug development with some early success with L-methionine and L-histidine as inhibitors (Jakubowski and Pick, 1983), and dexamethasone as an activator (Fingerle-Rowson et. al, 2003).
Figure 2: JMOL View of Human MIF
Source: Protein Data Bank
Inflammation Response:
MIF plays an essential role in the body’s inflammation response to infection. A general inflammation response basically proceeds as follows: After any penetration of our first line of defense, the epithelial surface, inflammation is induced with cytokines (including MIF) and chemokines released from tissue macrophages recognizing leaking plasma as well as bacterial products (such as proteins or carbohydrates, like LPS). These chemicals draw macrophages and dendritic cells into the area, as well as initiate blood clotting to prevent pathogen spread. The dendritic cells return to the lymph after antigen uptake in order to initiate the adaptive immune response. Additional macrophages arrive at the inflammation site through the process of extravasation. This process proceeds through migrating macrophages binding to cell surface adhesion molecules in the blood vessel lumen, which have been upregulated by cytokines. Next, the macrophage proceeds through the basement membrane of the vascular endothelium, in a process known as diapedesis, and arrives at the tissue site of inflammation (Janeway et. al, 2005).
The initial discovery by Bloom et. al (1966) that MIF inhibits macrophage migration provided insight into the protein’s role in localizing macrophages to sites of infection and inflammation. However, MIF has now been shown to have many more effects than just that; mainly working as a macrophage activator, but also playing other parts in both the innate and adaptive immune system, as well as the endocrine system.
MIF normally circulates in the blood at 2-4ng/mL. Macrophage cells and t-cells have been found to contain preformed pools of MIF (2-4 fg) so it can be rapidly secreted after exposure to endotoxins or cytokines such as TNF-α or some interferons. In fact, secretion and further translation is induced by levels of LPS 10-100 times lower than those needed to induce TNF-α (Tumor Necrosis Factor) production. As regulators of the inflammatory response, MIF and TNF-α have similar actions, both inducing inflammation and macrophage response to the point that their actions can be lethal, resulting in toxic shock syndrome. In addition, MIF and TNF-α induce each other; working together to stimulate an effective inflammatory response (Bucala, 1996).
By activating macrophages, MIF is not only increasing their rate of phagocytosis, but also inducing them to produce antimicrobial compounds, such as NO (Leng, 2003). MIF also sustains macrophage survival for an inflammation response by suppressing p53-dependent apoptosis. It does this by inhibiting the activation of the p53 tumor suppressor gene. This has important ramifications, which are discussed below, for cancer pathology (Mitchel et. al, 2002). MIF is also a survival signal for neutrophils, preventing release of BCL-2 compounds in the cell, and cytochrome c and smac from the mitochondria, which would activate caspases leading to cell death through a caspase signaling cascade (Bauman, 2003).
MIF plays other roles in the innate immune response, “functionally linking the cytokine network with the coagulation cascade during various wound healing processes. Because endothelial cells are a potential source of MIF, MIF may play an important role as a positive regulator within the whole process of wound repair” (Shimizu et. al, 2004). Lastly, MIF further contributes to the innate immune response by upping TLR-4 (toll-like receptor) concentration, leading to increased sensitivity to bacterial LPS (Roger, 2003).
MIF affects many systems in the body. For example, the sum of its inflammation effects (increased phagocytosis, sensitivity, etc.) leads to delayed-type hypersensitivity, t-cell priming, and antibody production by the adaptive immune response (Leng, 2003). Of course, the powerful local effects of MIF need to be kept in check by a negative feedback loop, and it has recently been shown that MIF is actually an integral part of the endocrine system, serving as a local mediator for the immune system against the effects of the endocrine system. Basically, “the physiologic role of MIF is to act at an inflammatory site or lymph node to counterbalance the profound inhibitory effects of [endocrine produced] steroids on the immune response” (OMIM, 2006).
Endocrine Role:
Glucocorticoids (anti-inflammatory hormones) are released as part of a systemic stress response by the pituitary gland. “Because glucocorticoids are an integral part of the host’s global response to infection or tissue invasion, the physiological role of MIF is to act at an inflammatory site or lymph node to counterbalance the profound inhibitory effects of steroids on the immune response” (Bucala, 1996). MIF is currently also known to be released from the anterior pituitary (making up about .05% of pituitary protein) to deal with systemic stress which encourages further MIF release by macrophages, t-cells, and eosinophils (Senter et. al, 2002). Increased release of MIF acts in an autocrine manner to overcome inflammatory mediator inhibition by glucocorticoids. In general, MIF “circulates at counterregulatory levels that increase during infection, inflammation, or stress. Secretion occurs systemically (pituitary) and locally (immune cells)” (Bucala, 1996). It is important to appreciate this dual role of MIF as a general endocrine compound and a specific immune compound. The importance of this feedback loop is best emphasized by the fact that “the MIF promoter has elements that may be sensitive to both proinflammatory (NF-kB, VK-1) and endocrine factors (nGRE, CREB)” (Bucala, 1996).
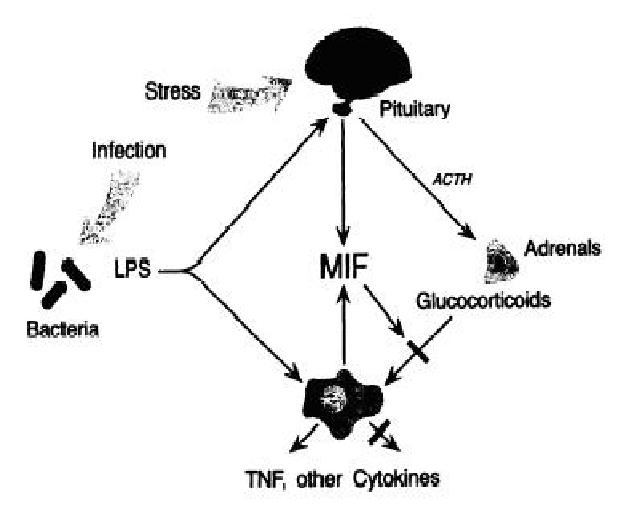
Figure 3: Diagram Summary of MIF's Endocrine and Immune System Interactions
Source: with permission from Bucala, 1996
Because of its inflammatory role, overexpression of MIF has been linked to toxic shock syndrome (sepsis), much like overexpression of TNF-α. In both cases, an effective immune system cytokine is utilized by the body too much, to the point where it induces so much fluid loss that it becomes life-threatening. Too strong a macrophage response can lead to this, as well as granulomatous disorders and autoimmune problems (Mitchel, 2002). It has been suggested that MIF inhibition may be of benefit for individuals with toxic shock (Donn and Ray, 2004). One potential route for doing this relies on lowering TLR4 expression by inhibiting PU1, a mouse transcription factor, and thus decreasing LPS sensitivity, which decreases MIF expression (Koebernick et. al, 2002). In mice, it has been demonstrated that MIF gene knockout does indeed give increased protection from toxic shock syndrome (Bucala, 1996).
As might be expected for an inflammatory mediator, MIF has been linked to arthritis. It is believed that MIF in synovial cells that is over stimulated by glucocorticoids can lead to chronic inflammation (Santos, 2001). Several gene targets have been identified that link MIF with arthritis. There is a promoter haplotype that is linked to adult inflammatory arthritis (Donn and Ray, 2004). Researchers have also found a particular gene polymorphism at position 173 that demonstrates an increased risk for systemic juvenile rheumatoid arthritis. This mutation has been shown to create an activator protein binding site, leading to increased serum and synovial fluid levels of MIF, and thus a propensity to develop arthritis (OMIM, 2006). A tetranucleotide repeating sequence has been found at position 794 that leads to higher gene transcription, correlating with arthritis severity (Baughe et. al, 2002).
The logical treatment for MIF associated arthritis is blockage of MIF with antibodies, and this indeed has been shown to delay the onset of arthritis. However, since MIF is necessary to promote Th1 response you can’t block it too much with out running into negative immunological effects. For example, MIF knockout mice have increased susceptibility to Salmonella typhimurium, demonstrating the difficulty in balancing MIF reduction and increased pathogen susceptibility (Koebernick et. al, 2002).
Recently, MIF has been associated with cancer development. One of the ways in which MIF sustains macrophage survival is by suppressing, through inhibition, the p53 apoptosis pathway. Exposure to LPS starts macrophages down this apoptotic pathway that eventually finishes with the caspase signal cascade (Mitchel et. al, 2002). Findings that MIF is overexpressed in tumors encouraged researchers to further investigate MIF's role in tumor development (Takakishi, 1998). In 2003, it was shown that MIF, by inhibiting p53 gene activation and thus cellular apoptosis can, at sites of chronic inflammation, inhibit p53 to the point that genetically damaged cells are not kept in check, allowing cell proliferation that can eventually lead to tumors (Fingerle-Rowson et. al, 2003). MIF has additionally been shown to induce p27 expression, a growth protein, also having implications for cancer development (Kleeman et. al, 2000)
Sources:
Baumann et. al. Macrophage migration inhibitory factor delays apoptosis in neutrophils by inhibiting the mitochondria-dependent death pathway. FASEB Journal. 17: 2221-2230, 2003.
Baughe et. al. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 3: 170-176, 2002.
Bloom, B.R. and Bennet, B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science 153: 80-82, 1966.
Bucala, R. MIF rediscovered: cytokine, pituitary hormone, and glucocorticoid-induced regulator of the immune response. FASEB Journal. 10: 1607-1613, 1996.
Donn, R.P., Ray, D.W. Macrophage migration inhibitory factor: molecular, cellular and genetic aspects of a key neuroendocrine molecule. Journal of Endocrinology. 182: 1-9, 2004.
Fingerle-Rowson et. al. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc. Natl. Acad. Sci. 100: 9354-9359, 2003.
Fingerle-Rowson et. al.Regulation of macrophage migration inhibitory factor expression by glucocorticoids in vivo. Am J Pathol. 162: 47-56, 2003.
Janeway, et. al. Immunobiology. Garland Science: New York, NY. 2005. 50-61.
Jakubowski A, Pick E. Modulation of the action of macrophage migration inhibitory factor by antioxidants and drugs affecting thromboxane synthesis. Immunopharmacology 6:215-29, 1983.
Kleeman et. al. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature. 408: 211-216, 200.
Koebernick et. al. Macrophage migration inhibitory factor (MIF) plays a pivotal role in immunity against Salmonella typhirium. Proc. Natl. Acad. Sci. 99: 13681-13686, 2002.
Leng et. al. MIF Signal Transduction Initiated by Binding to CD74. J. Exp. Med. 197: 1467-1476, 2003.
Lolis Labs Home Page: <http://info.med.yale.edu/pharm/Lolis/index.html>. Accesed on March 10, 2006.
Mitchell et. al. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: Regulatory role in the innate immune response. Proc. Natl. Acad. Sci. 99: 345-350, 2002.
Nguyen et. al. A 16-Residue Peptide Fragment of Macrophage Migration Inhibitory Factor, MIF-(50–65), Exhibits Redox Activity and Has MIF-like Biological Functions. J. Biol. Chem. 278: 33654-33671, 2003
Online Mendelian Inheritance in Man: Macrophage Inhibiton Factor. <http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=153620>. Accessed on March 10, 2006.
Protein Data Bank: <http://www.pdb.org/pdb/static.do?p=explorer/viewers/jmol.jsp>. Accessed on March 14, 2006.
Roger, et. al. Macrophage migration inhibitory factor (MIF) regulates host responses to endotoxin through modulation of Toll-like receptor 4 (TLR4). J. Endotoxin Research. 9: 119-123, 2003.
Santos et. al. Role of macrophage migration inhibitory factor (MIF) in murine antigen-induced arthritis: interaction with glucocorticoids. Clin. Exp. Immunol . 123, 309-314, 2001.
Senter et. al. Inhibition of macrophage migration inhinitory factor (MIF) tautomerase and biological activities by acetaminophen metabolites. Proc. Natl. Acad. Sci. 99: 144-149, 2002.
Shimizu et. al. Macrophage Migration Inhibitory Factor Is Induced by Thrombin and Factor Xa in Endothelial Cells. J. Biol. Chem. 279: 13729-13737, 2004.
Takahashi et. al. Involvement of macrophage migration inhibitory factor (MIF) in the mechanism of tumor cell growth. J. Mol. Med. 11: 707-714, 1998.
Send comments, questions, and suggestions to: mawilson@davidson.edu