Introduction:
Psoriasis is an autoimmune disease that presents on the skin as itchy, thick, red, inflamed plaques with silvery scales. Clinically, psoriasis is characterized by hyper-proliferation of basal keratinocytes (skin cells) leading to a thickened, scaly, epidermis (Bowcock, 2005). Essentially, affected skin cells rapidly pile up on the skin surface instead of fully differentiating. This rapid turnover is triggered by T-cells and local inflammation, encouraging keratinocytes to replicate every 3-4 days instead of the standard 20-30 days. Psoriasis equally affects males and females and it typically develops on the elbows, knees, back, butt, scalp, palms, or the soles of the feet, but can form anywhere including under nails, in the mouth, and on genitalia. The disease onset is usually before the age of 35. Psoriasis presents in several forms: plaque, guttate (triggered by respiratory infection), pustular, inverse (smooth patches), erythrodermic (widespread), and as psoriatic arthritis (developed by 30% of psoriatic individuals) (NIAMS, 2006). Figures 1 and 2 (below) show some of these various types of psoriasis.
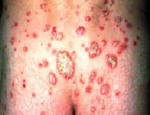
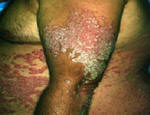
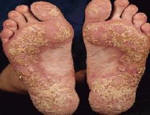
Figure 1: Images of guttate psoriasis, psoriasis on the body, and psoriasis on the feet.
Source: http://www.psoriasiscafe.org/psoriasis-pictures.htm
Psoriasis has been clinically described for 2000 years and it presently affects 2-3% of the world’s population. Despite the numerous advances in our understanding of the disease, the direct cause of psoriasis remains indeterminate. It is still unclear if psoriasis is the result of a mutation in keratinocytes that causes abnormal immune proliferation or if it is instead due to a mutation in immunocytes that leads to the proliferation (Nickoloff, 1999). Much of the recent progress in the field has come from linkage analysis of diseased genotypes, as it is known that one-third of cases have a family history of psoriasis. However, environmental triggers, such as streptococcal/superantigens, injury, stress/neuropeptides, lithium and beta-blocker use, smoking, and alcohol use are equally important in the development of psoriasis (Bowcock, 2005). The development of psoriasis is also associated with arthritis, as well as AIDS, where it can present with particularly severe plaques (Tomfohdre, 1994). Recent progress in our understanding of the effects of T-cells at locally inflamed psoriatic plaque sites is contributing to the development of several new targeted treatments. Please keep in mind that psoriasis, with its painful and unsightly sores, can be particularly psychologically traumatic, further contributing to the condition’s effect on a patient’s quality of life.
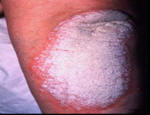
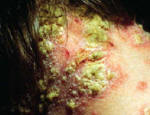
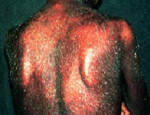
Figure 2: Images of psoriasis on the knee and pustular and erythrodermic psoriasis.
Source: http://www.psoriasiscafe.org/psoriasis-pictures.htm
Autoimmune Response:
Psoriasis results from an autoinflammatory immune response. It is still unknown exactly what is inciting the response, but the mechanisms behind disease pathogenesis are well understood. Epidermal autoantigens (self-proteins in the skin that would serve to incite the initial immune response) for psoriasis have not yet been conclusively identified. However, because T-cells are known to selectively expand and localize in response to developing psoriatic plaques it is suspected that there most be some sort of autoimmunogenic protein that either genetically, environmentally, or stochastically initiates clonal-expansion either through its own mutation or through an autoreactive T-cell receptor (TCR) that escaped detection during development. Currently, some candidates for possible autoantigens are keratin 13, Heterogeneous Nuclear Ribonucleoprotein-A1, and Rab Coupling Protein Isoform (Bowcock, 2005). However, no common CDR (complementary determining region) motifs have been found in comparative studies so there is quite possibly various autoantigens interacting with the TCR in different patients (Chang, 1994). It has been particularly difficult to figure out the interactions of auto-reactive T-cell conditions, like psoriasis, because, due to their MHC restriction and the small peptide fragment they recognize, autoreactive T-cells can be more difficult to investigate than autoantibodies (Janeway, 2005).
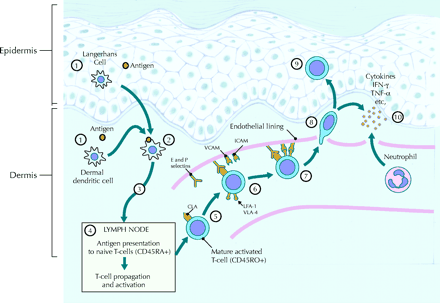
Figure 3: Initiation of a psoriatic immune response with antigen presenting cells delivering an as yet
unknown autoantigen to T-cells, leading to cytokine production and disease pathogenesis.
Source: http://www.cmaj.ca/cgi/content-nw/full/170/13/1933/F427
In active psoriatic plaques, research has found that CD8+ T-cells predominate in the epidermis of psoriatic lesions, while CD4+ T-cells are found in the dermis; Langerhans’ cells and macrophages are also recruited to the diseased area (Chang, 1994). The localized skin damage and rapid cell turnover that occurs in psoriasis is the result of the standard CD8+ attack response, with selective exocytosis of perforin, granulysin, and granulozymes; chemicals which lyse and induce apoptosis in targeted cells. Activated CD8+ cells have no need for co-stimulation and will continue proliferating and attacking as long as they are in the presence of antigen. Other responding cells serve to further incite cytokine production leading to increased inflammation. One particular cytokine, IL-6, is heavily overexpressed in psoriatic plaques and functions as a lymphocyte growth factor (Grossman, 1989). The runaway cytokine production of Th1 cells in psoriatic plaques leads to increased inflammation and cell-recruitment for a continued autoreactive positive feedback response loop which can lead to epitope spreading (development of additional autoreactive lymphocytes because of a continuous attack response). Some particularly important mediators which have become potential therapeutic targets are, for example, TNF-α, IFN-γ, IL-1, and CDSN (a cell-adhesion cytokine) (Bowcock, 2005). Further demonstrating the importance of cell recruitment to disease progression was a study by Bullard et. al (1996) demonstrating that sub-lethal mutations in the leukocyte adhesion molecule, CD-18, could lead to psoriasis like skin disease in mice.
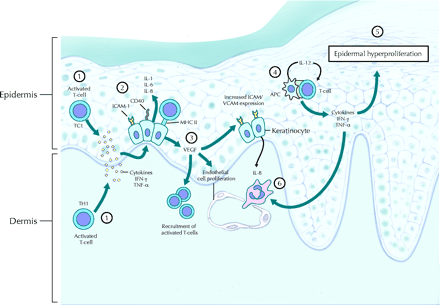
Figure 4: Localized immune response, by cytotoxic T-cells responding to cytokine
production, against keratinocytes in an active psoriatic plaque.
Source: http://www.cmaj.ca/cgi/content-nw/full/170/13/1933/F627
It has been estimated that as many as 30% of psoriasis patients develop psoriatic arthritis, where autoreactive CD8+ T-cells, just like for psoriasis, accumulate in afflicted synovial fluid causing joint deterioration. Psoriatic skin involvement generally precedes joint-disease and patients need to be monitored for the development of this syndrome. The main difference between a skin psoriatic response and that in a joint is in the different homing receptors (CLA, Cutaneous Lymphocyte Associated antigen is the one required for skin) (Cassell, 2005).
Linkage studies have revealed many genes potentially associated with psoriasis. There are multiple psoriasis susceptibility loci, some are suspected on chromosomes 17 and 19, but most of the linkage is associated with MHC I alleles on chromosome 21. HLA-Cw6 is one possible site, as well as another HLA locus termed PSORS1. Within PSORS1 there are at least 2 genes with haplotypes suspected to be associated with psoriasis susceptibility. HCR (α-helix coiled rod homolog) is overexpressed by keratinocytes in psoriatic lesions and can lead to upregulation of cytokeratins and, eventually, psoriasis. CDSN is another gene found in the PSORS1 locus. It is involved with cell adhesion and is upregulated by TNF-α. The protein products of these genes were found to be overexpressed in psoriatic lesions (Bowcock, 2005).
Brian J. Nickoloff (1999) proposes a very interesting explanation for the prevalence of psoriasis in the world population (2-3%). Basing his arguments in evolutionary immunology, Nickoloff proposes that psoriasis is an evolutionary relic of selection to strengthen the “shield” of our stratum corneum (outer skin layer), which is our primary protection from infections. In his view, psoriasis is seen as one end of the spectrum of selection encouraging us to be able to respond to trauma or infection of the skin with “an exaggerated T-cell response [that produces] a psoriatic plaque, which solves the primordial problem of controlling the spread of dangerous pathogens.” The unfortunate end of this selection is the formation of plaques that can last for decades. Psoriatic plaques lack the skin’s normal stratum corneum because of abnormal differentiation, but are even more resistant to bacterial invasion. This cutaneous hyperresonsiveness could be seen as an “early-warning back up system” that utilizes both the innate and adaptive branches of the immune system. Nickoloff cites several research findings as evidence, emphasizing the importance of what causes the psoriatic response to be initiated by the immune system: Unrelated skin lesions often form following infections and spread as a result of Th1 cytokine production. Intradermal injection of IFN-γ, stimulating an infectious response, can produce psoriatic lesions in susceptible patients. Plaques often form in response to trauma, such as the Koebner response, where tape is ripped off of the skin leading to a psoriatic plaque stripe. Lastly, Nickoloff cites that plaques resist apoptosis as well as transformation, making a great immunological barrier, which can even withstand exposure to coal tar, UV radiation, and immunosuppressive drugs, which, unfortunately for us patients, are the standard treatments for psoriasis.
Psoriasis is usually treated with topical steroids, then phototherapy, then systemic immune therapy. Corticosteroids reduce inflammation, skin-cell turnover and local immune response. Local vitamin D3 can also slow skin cell turnover. Topical pealing agents, such as salicylic acid and coal tar, can be used to reduce the surface plaque. Combining local treatment with UV-therapy can be even more effective. Some systemic treatments include Methotrexate, retinoids (most effective for pustular psoriasis), Cyclosporine, and other drugs that suppress the immune system and, particularly, the psoriatic T-cell response. Unfortunately, plaques become resistant to treatment over time and thus, treatments should rotate. It is important to treat psoriasis early before plaques are widespread; particularly considering the process of epitope spreading (explained above), which is suspected to contribute to worsening psoriasis. One of the newer therapies used to treat psoriasis has shown positive results. A progressive dose elevation of IL-4 induced Th2 differentiation, which can be measured by the resulting decrease in inflammatory cytokine concentration, leading to psoriasis remission in some patients (Ghoreschi, 2003). This therapy works by slowly shifting the T-cell response from an inflammatory-cytokine producing Th1 response to a Th2 response that, although negatively associated with allergies, does not encourage the harmful CD8+ T-cell response seen in psoriasis. Similar treatments are being pursued using IL-10 and IL-11, also Th2 promoting cytokines. Additionally, an IL-12 blocker, which would inhibit Th1 response, has just entered into clinical trials (Winterfield, 2005). Many of the therapies discussed above are also helpful in treating psoriatic arthritis (Cassell, 2005).
Numerous targeted therapies for psoriasis have recently entered into clinical trials. Seeing as Methotrexate and Cyclosporin are inadequate for long term use due to their immunosuppressive effects, new targeted biological agents, such as Alefacept, Efalizumab, and Etanercept have been developed. Alefacept is a “fusion-protein” consisting of LFA-3 (Leukocyte Function Associated Antigen) with the Fc receptor of IgG. It blocks CD2 on T-cells and prevents co-stimulation, and thus subsequent activation, by binding its LFA fragment on Antigen presenting cells. Alefacept also initiates apoptosis in memory cells expressing high levels of CD2. One dangerous side effect of this drug is a drop in the CD4+ cell count. Efalizumab is an anti-CD11a antibody that blocks LFA-1 binding, preventing activation and migration from the blood vessels to the skin. Etanercept is an anti-TNF-α receptor protein which is used to modulate the local immune response. Unfortunately, many of these drugs are immunosuppressives to an extent also and, additionally, are immunogenic themselves (Winterfield, 2005).
Sources:
Bowcock, A.M. The Genetics of Psoriasis and Autoimmunity. Annu. Rev. Genomics Hum. Genet. 6: 93-122, 2005.
Bullard et. al. A Polygenic Mouse Model of Psoriasiform Skin Disease in CD-18-Deficient Mice. Proc. Nat. Acad. Sci. 93: 2116-2121, 1996.
Cassell, S. and Kavanaugh, A. Psoriatic arthritis: Pathogenesis and novel immunomodulatory approaches to treatment. J. Immune Based Therapies and Vaccines. 3: 6, 2005.
Chang et. al. CD8+ T Cells in psoriatic lesions preferentially use T-cell receptor V β 3 and/or V β 13.1 genes. Proc. Nat. Acad. Sci. 91: 9282-9286, 1994.
Ghoreschi et. al. Il-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nature Medicine. 9: 40-46, 2003.
Grossman et. al. IL-6 is expressed in High Levels in Psoriatic Skin and Stimulates Proliferation of Cultured Human Keratinocytes. Proc. Nat. Acad. Sci. 6: 6367-6371, 1989.
Janeway, et. al. Immunobiology. Garland Science: New York, NY. 2005.
National Institute of Arthritis and Muscoskeletal and Skin Diseases Psoriasis Summary: <http://www.niams.nih.gov/hi/topics/psoriasis/psoriasis.htm> Accessed on April 18, 2006.
Nickoloff, B. Skin Innate Immune System in Psoriasis: friend or foe? J. Clinical Investigation. 104: 1161-1164, 1999.
“ Psoriasis and the new biologic agents: interrupting a T-AP dance” — Reprinted from, CMAJ 22-Jun-04; 170( 13), Page(s) 1933-1941 by permission of the publisher. © 2004 CMA Media Inc.
Psoriasis Cafe: <http://www.psoriasiscafe.org/psoriasis-pictures.htm>. Accessed on April 20, 2006.
Tomfohdre et. al. Gene for Familial Psoriasis Susceptibility mapped to the Distal End of Human Chromosome 17q. Science. 264: 1141-1145, 1994.
Winterfield et. al. Psoriasis Treatment: current and emerging directed therapies. Ann Rheum Dis. 64: 87-90, 2005.
Send comments, questions, and suggestions to: mawilson@davidson.edu