MacDNAsis of Human Cytochrome p450 Reductase
Introduction: Using the MacDNAsis software on the cDNA and
amino acid sequence of human cytocrome reductase, I furthered my
understanding of this protein. First, the MacDNAsis predicted the open
reading frame of the human (Homo sapien) cytochrome reductase cDNA.
Then, the molecular weight, hydropathy, antigenicity, and secondary structure
were predicted. Finally, the cDNA of human cytochrome reductase was
compared to four other cytocrome reductase cDNAs (from
Genebank) to determine the percent similarity of the five sequences.
1. Open reading frame determination:
MacDNAsis utilized the cDNA of human cytochrome
reductase to determine the possible open reading frames (orf).
The largest open reading frame, highlighted in black (Fig. 1), was assumed
to be the coding region of the human cytochrome reductase. The reading
frame is contained within the amino acids numbered 544 and 2028.

Figure 1: MacDNAsis open reading frame analysis of human cytochrome
reductase. The three different rows indicate the three different
reading frame possibilities of the cDNA sequence. The start codons
are represented by the red triangles, and the green lines represent the
stop codons. The white areas between the triangles and the lines
indicate open reading frames. The largest reading frame was assumed
to be the coding region for the human cDNA which is highlighted black.
2. Molecular Weight Determination:
After MacDNAsis determined the orf, the molecular weight was calculated
after translating the cDNA into the appropriate amino acids sequence of
cytochrome reductase. The analysis predicted the molecular weight of cytochrome
reductase to be 56672.8 daltons.
3. Determination of Cytochrome reductase hydropathy:
Using a Kyte and Doolittle plot, the hydrophobic and hydrophilic regions
of the human cytochrome reductase protein were determined (fig 2).
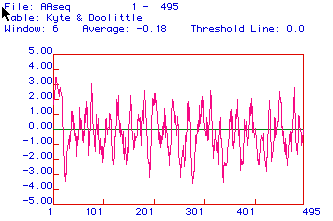
Figure 2: Kyte and Doolittle analysis determined hydrophobic
and hydrophilic regions of the coding sequence of human cytochrome reductase.
Positive values on the x-axis, indicate hydrophobic regions while negative
values indicate regions that are hydrophilic. The y-axis indicates
the amino acids corresponding to the different hydrophobic and hydrophilic
regions. Amino acid segments that are determined greater than 2, suggest
possible integral membrane proteins. Cytochrome reductase has many
hydrophobic regions, with the greatest hydrophilic region at 1-20 amino
acids which could be a section of the protein that spans the inner mitochondria
membrane(Fig. 2).
4. Hopp and Woods Antigenicity Plot of Cytochrome reductase:
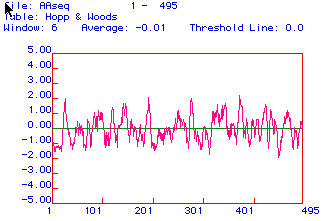
Figure 3: Hopp and Woods antigenicity plot of cytochrome reductase
predicts areas that are hydrophilic. Hydrophilic regions which are
above the x-axis, are positive and therefore might be very antigenic.
Cytochrome is extremely variable, but the region between 325-360 amino
acids has high antigenicity potential and could be utilized to generate
a peptide to make a monoclonal antibody against (Fig 3).
5. Determination of Secondary Structure:
Next, the secondary structure of human cytochrome reductase
was predicted to help visualize the structure of the protein. The
secondary structure of a protein can be predicted by considering the likely
interactions of the amino acid side chains.
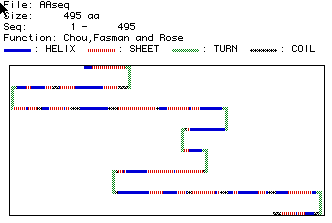
Figure 4: This analysis predicts the secondary structure of human
cytochrome reductase. The blue bars represent alpha helixes, red
striped bars represent beta strands, green bars represent turns in the
structure, and black striped bars indicate coiled regions. This analysis
indicates that human cytocrome reductase includes; about 16 alpha helixes,
23 beta strands, 7 turns in the structures, and 10 coiled regions.
This prediction can be compared to a similar Rasmol
image of a cytochrome reductase.
6. Multiple Sequence Alignment:
Using Higgins analysis, MacDNAsis compared five cytochrome reductase
amino acid sequences from five species; including, Homosapien,
Saccharomyces cerevisiae, Mus
musculus, Drosophila melanogaster, and
Arabindopsis thaliana. The sequences were
provided by genebank, see my genebank search.

Figure 5: This analysis compares the amount of sequence similarity
that exists between five sequences of cytochrome reductase. The line labeled
human represents the amino acid sequence for Homo sapien. The line
labeled mouse represents the amino acid sequence for Mus musculus.
The line labeled plant represents the amino acid sequence for Arabindopsis
thaliana. The line labeled fly represents the amino acid sequence for
Drosophila melanogaster. The line labeled yeast represents the amino
acid sequence for Saccharomyces cerevisiae. Letters represent the
one letter abreviations for the amino acids. The highlighted amino acids
represent similar amino acids as compared to the other sequences listed.
The small horizontal lines in the sequences signify spaces in certain species
that helped maximize sequences alignment. The numbers on the left,
right, and above the sequences represent the corresponding amino acids.
As seen in the figure, similarity has been well conserved between the human
and mouse for the sequence corresponding to amino acids 40 - 50 (fig 5).
7. Phylogenetic Tree Analysis:
This phylogenetic tree of cytochrome reductase was assembled using
the analysis of similarity determined from the sequence alignment above
(fig 5).
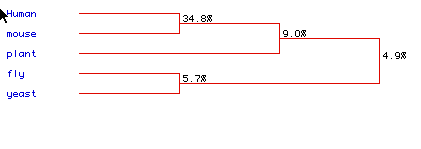
Figure 6: Phylogenetic tree comparing the similarity in amino acid
sequence of the five species; Homosapien,
Saccharomyces cerevisiae, Mus
musculus, Drosophila melanogaster, and
Arabindopsis thaliana. The percentages
indicate the amount of similarity between the sequences. The human and
mice sequences have 34.8% similar amino acid sequences (Fig 6).
While only a 4.9% similarity was determined between all five sequences.
Evolutionary lineages can be predicted from these trees because people
assume amino acid similarity can suggest conservation of amino acid sequence
through evolution. Therefore, sequences that are less similar hypothetically
diverged from the other sequences the earliest, or their mutation rate
is quicker. Following this theory, this tree shows that yeast and
fly share a more conserved cytochrome sequence than do flies and humans.
However, little similarity was found between any of the structures suggesting
that each sequence cytochrome reductase has changed significantly from
the other species over time.
Return to my Home page
Return
to Davidson College Biology Department Home Page
Send comments, and suggestions to: mocamp@davidson.edu