{kind=link}

Cell, Vol. 48, 899-907, March 13, 1987, Copyright © 1987 by Cell Press
Sean Munro and Hugh R. B. Pelham
MRC Laboratory of Molecular Biology
Hills Road,
Cambridge CB2 2QH,
England
Summary
Proteins that permanently reside in the lumen of the endoplasmic reticulum (ER) must somehow be distinguished from newly synthesized secretory proteins, which pass through this compartment on their way out of the cell. Three luminal ER proteins whose sequence is known, grp78 ("BiP"), grp94, and protein disulphide isomerase, share the carboxy-terminal sequence Lys-Asp-Glu-Leu (KDEL). We show that deletion (or extension) of the carboxyl terminus of grp78 results in secretion of this protein when it is expressed in COS cells. Conversely, a derivative of chicken lysozyme containing the last six amino acids of grp78 fails to be secreted and instead accumulates in the ER. We propose that the KDEL sequence marks proteins that are to be retained in the ER and discuss possible retention mechanisms.
Introduction
The lumen of the ER contains several proteins that appear to be soluble rather than membrane-bound. The most prominent of these are the glucose-regulated proteins grp78 and grp94, so called because their synthesis is stimulated when fibroblasts are deprived of glucose (for review see Pelham, 1986). We have recently reported the complete cDNA sequence of grp78 and have shown that it is related to the heat shock protein hsp70 and is identical with "immunoglobulin heavy chain binding protein" (BiP) (Munro and Pelham, 1986; Haas and Wabl, 1983; Bole et al., 1986). Grp94 is a glycoprotein that is related to another heat shock protein, hsp90 (Sorger and Pelham, 1987). It is one of the most abundant proteins of the ER and has been variously referred to as grp94 (Shiu et al., 1977; Lee et al., 1984), "100K" (Welch et al., 1983), "hsp108" (Kulomaa et al., 1986; see also Sorger and Pelham, 1987), "endoplasmin" (Koch et al., 1986), and "ERp99" (Lewis et al., 1985; M. Green, personal communication). Other proteins in the lumen include protein disulphide isomerase (Freedman, 1984; Edman et al., 1985) and prolyl-4-hydroxylase, one of the enzymes that modifies newly synthesized collagen molecules in the ER (Kivirikko and Myllyla, 1982).
These proteins occupy the same functional compartment as newly synthesized secretory proteins, but do not share their fate. A clear example is that of grp78 (BiP), which forms a complex with newly synthesized immunoglobulin heavy chains. Subsequently, the heavy chains associate with light chains and are secreted, while grp78 remains in the ER (Bole et al., 1986). There must therefore exist a mechanism by which the cell distinguishes resident luminal ER proteins such as grp78 from secreted proteins such as immunoglobulins. In principle, this could involve recognition of either a retention signal or a secretory signal on the appropriate proteins.
We have previously noted that the three soluble ER proteins whose sequences are known (grp78, grp94, and protein disulphide isomerase) share a common carboxyterminal tetrapeptide sequence (Munro and Pelham, 1986; Sorger and Pelham, 1987). We show here that the carboxyl terminus of grp78 is crucial for its retention in the ER and that transfer of the last six amino acids to a secreted protein, chicken lysozyme, is sufficient to cause retention of lysozyme in the ER.
Results
An Intact Carboxyl Terminus Is Necessary for Retention of grp78 in Cells
Figure 1. Comparison of Amino- and CarboxyTerminal
Sequences of ER Proteins
The sequences shown are from rat grp78 (Munro and Pelham, 1986),
rat protein disulphide isomerase (P.D.I.; Edman et al., 1985),
hamster grp94 (Sorger and Pelham, 1987), and chick grp94 (Kulomaa
et al., 1986; these authors refer to the protein as hsp108, but
comparison with the hamster sequence identifies it as grp94).
For the grps, the sequences shown are from those regions that
are not homologous to heat shock proteins. Charged residues are
indicated by + or - above the sequence, and the conserved carboxy-terminal
tetrapeptide is boxed. The one-letter amino acid code is used.
The sequences of grp78, grp94, and protein disulphide isomerase show similarities at both their amino and carboxyl termini, although the proteins are otherwise quite different from each other. In the case of the grps, the similarities lie outside the regions that are homologous to the heat shock proteins (Figure 1). We reasoned that one or both of these terminal sequences might be important for retention of the proteins in the cell. To test this idea, we deleted portions of a rat grp78 cDNA clone and inserted the altered coding sequences into a COS cell expression vector. The amino acid sequences at each end of the mutant proteins are indicated in Figure 2. In the first mutant (SAGM2), the carboxy-terminal 60 amino acids of the protein were replaced with an 11 amino acid sequence derived from the human c-myc gene. The myc sequence was chosen because it is recognized by an existing monoclonal antibody and thus forms a convenient tag for detection of the expressed protein. In another mutant (SIG2), the leader peptide and first 11 amino acids of grp78, which are not homologous to hsp70, were replaced with the leader peptide and first six amino acids of an immunoglobulin heavy chain. A control plasmid (SAG1) encodes intact grp78. These constructs were transfected into COS cells, and 45 hr later the cells were labeled with [35S]methionine for 2 hr. Labeled proteins in the cells and culture supernatants were then analyzed by SDS-polyacrylamide gel electrophoresis and fluorography.

Figure 2. Terminal Sequences of grp78 and Mutants
Plasmid names are at the left, and the relevant amino acid sequences
are shown in the one letter code. Slashes indicate junctions between
grp78 sequences and others. The KDEL sequence is underlined. The
sequence EQKLISEEDL, present in SAGM2, SAGM6, SIGM2, and SAGMK1,
is derived from the human c-myc gene and is recognized
by monoclonal antibody 9E10. Numbers refer to residues of the
mature wild-type grp78 protein.
Transfection of cells with the wild-type construct (SAG1) produced no new labeled protein in the culture supernatant relative to the untransfected control (Figure 3A). The transfected cells, however, contained an abundance of labeled grp78 that could be immunoprecipitated with a polyclonal antibody raised against the purified rat protein (Figure 3B). This antibody also recognized the endogenous monkey grp78, but the amounts synthesized in untransfected COS cells were much lower than in the transfected cells. We were able to quantitate the accumulation of grp78 cells by probing protein blots with a monoclonal antibody (7.10) that recognizes most hsp70-related proteins (see Munro and Pelham, 1986). Such experiments showed that the transfected cell population contained about 3-fold more grp78 than untransfected cells (data not shown). Since the transfection efficiency (measured by immunofluorescence) was typically 20%40%, individual cells must have contained at least 7-fold more grp78 than normal. Despite this considerable overproduction, the protein was efficiently retained within the cells.
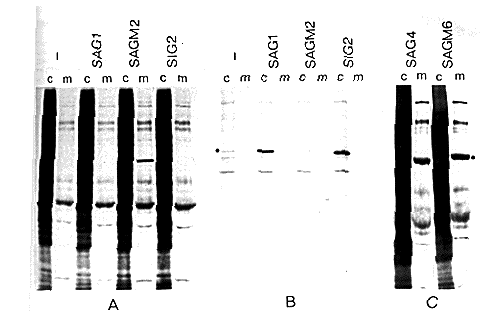
Figure 3. Expression of Amino- and Carboxy-Terminal
Deletion Mutants of grp78
(A and C) COS cells that were untransfected (-) or transfected
with the indicated plasmid were labeled with [35S]Met
for 2 hr, and samples of the cells (c) and culture medium (m)
were analyzed by SDS polyacrylamide gel electrophoresis and fluorography.
(B) Samples from the same experiment as (A) immunoprecipitated
with a polyclonal anti grp78 serum; equivalent amounts of the
cell and medium samples were analyzed. Dots indicate the position
of migration of grp78. SAG1 expresses wild-type protein; SAGM2,
SAG4, and SAGM6 are carboxy-terminal deletions, and SIG2 is an
amino-terminal deletion. The SAGM2 product is not recognized by
the polyclonal antiserum.
The amino-terminal deletion mutant SIG2 behaved in the same way as the wild-type protein, being present in the cells but absent from the medium (Figures 3A and 3B). In contrast, transfection of cells with the carboxy-terminal deletion mutant SAGM2 resulted in a new labeled band in the culture supernatant with the expected mobility of the truncated grp78 (Figure 3A). The altered protein was not recognized efficiently by the polyclonal antiserum (Figure 3B), but subsequent experiments with a monoclonal antibody identified this band as the product of SAGM2 (see below). Similar results were obtained with two other carboxy-terminal deletion mutants of grp78 (SAGM6 and SAG4), both being secreted from transfected COS cells (Figure 3C). These mutants lack only 15 amino acids from the grp78 sequence; in SAG4 they are replaced with the dipeptide Leu-Asp, while SAGM6 contains the c-myc sequences in addition (see Figure 2). Thus a variety of changes to the carboxyl terminus of grp78 have the effect of converting the protein from a resident of the ER to a secretory protein.
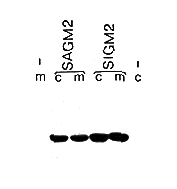
Figure 4. Blot Analysis of Expressed Proteins
Cells (c) or medium (m) from untransfected (-) or transtected
cultures were analyzed by protein blotting. Expressed proteins
were detected with the anti-myc monoclonal antibody 9E10.
SAGM2 has the carboxyl terminus deleted, while SIGM2 has both
the amino and carboxyl termini removed.
Figure 4 shows the effect of simultaneously removing both the amino and carboxyl termini of grp78. In this experiment, the mutant proteins were detected by protein blotting, using the monoclonal antibody that recognizes the c-myc peptide tag. The proportion of the expressed protein that reached the medium was the same, whether both termini were deleted (SIGM2) or only the carboxyl terminus (SAGM2). From this and the previous results, we conclude that the amino terminus has little effect on the retention of grp78 in the cell.
Experiments such as that shown in Figure 4 give an indication of the efficiency with which the carboxy-terminal deletion mutants are secreted. From such experiments, we estimate that an amount equivalent to the steady state intracellular level of the mutant SAGM2 is released into the supernatant in 4-5 hr, giving an approximate half-life in the cell of 3 hr for this protein. Similar half-lives were obtained for the other secreted mutants. Although quite slow, this rate of release is within the range observed for proteins that are secreted via the constitutive pathway (Gebhart and Ruddon, 1986).
The behavior of the three carboxy-terminal mutants indicates that deletion of part of grp78 is sufficient to cause its secretion. This strongly suggests that retention of the intact protein in the ER is an active process, requiring recognition of a specific structural feature. Conversely, the successful negotiation of the secretory pathway by the truncated proteins supports the idea that transit from the ER to the outside of the cell requires no special signal, since there is no reason why grp78 should contain such a signal.
The Last Six Amino Acid Residues of grp78 Contain an Important
Signal
Although the above experiments show that removal of 15 amino
acids from grp78 causes its secretion, they do not prove that
the putative retention signal is within that sequence; the deletions
may simply disrupt the folding of an entire carboxy-terminal domain.
However, comparison of the four luminal ER proteins whose sequence
is known shows that they have an identical carboxy-terminal tetrapeptide
LysAsp-Glu-Leu (KDEL in the one letter code) but diverge further
into the protein (Figure 1). To test whether it is the presence
of this conserved sequence that is important for retention, we
made a construct (SAGMK1) in which the last six amino acids of
grp78 were added back to the 60 amino acid deletion mutant, with
the c-myc peptide present at the junction (Figure 2). The
protein expressed from this construct was not secreted into the
medium (Figure 5A: SAGMK1), and probing of a protein blot with
the anti-myc monoclonal antibody confirmed that it remained
intact in the cells (Figure 5B). This strongly suggests that the
putative signal is encoded in the carboxy terminal hexapeptide
and does not require an elaborate tertiary structure to be recognized.
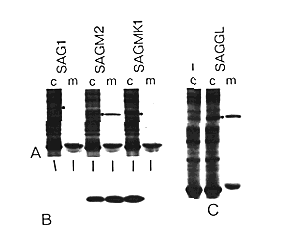
Figure 5. Requirements for Retention of grp78
in Cells
(A and C) ) [35S]Met labeling. SAG1 is wild type, SAGM2
lacks 60 amino acids from the carboxyl terminus, and SAGMK1 is
the same as SAGM2 but with the last 6 residues added back to the
carboxyl terminus. SAGGL encodes grp78 with a two-amino-acid extension
at the carboxyl terminus. Dots indicate the expressed proteins.
(B) Blot analysis of samples like those in (A) using the anti-myc
monoclonal antibody. Lanes correspond to those above them, as
indicated. SAG1 lacks the myc sequences and thus serves
as a control.
We next asked whether the conserved sequence had to be present at the extreme carboxyl terminus. The construct SAGGL encodes the entire grp78 sequence, but with a two-residue extension (Gly-Leu) at the carboxyl terminus (Figure 2). This protein was secreted from COS cells with about the same efficiency as the carboxyterminal deletion mutants (Figure 5C). Thus the presence of the conserved sequence at an internal position is not sufficient for retention; to be recognized, the signal must be at the carboxyl terminus. Moreover, since the extended protein has the same carboxy-terminal residue as grp78 (leucine), we can conclude that this residue itself is not a sufficient signal.
Addition of a Hexapeptide Sequence to Lysozyme Prevents
Its Secretion
Soluble ER proteins share features other than their carboxy-terminal
sequences; for example, the proteins mentioned in the introduction
are all quite strongly acidic. To see whether the carboxyl terminus
alone is sufficient to prevent secretion, we made a series of
constructions based on the chicken lysozyme gene. Lysozyme is
a small, highly basic protein that is constitutively secreted
from cells (Cutler et al., 1981; Kondor-Koch et al., 1985). If
the KDEL sequence is a sufficient retention signal, addition of
these amino acids to lysozyme should cause it to accumulate in
the ER rather than be secreted.
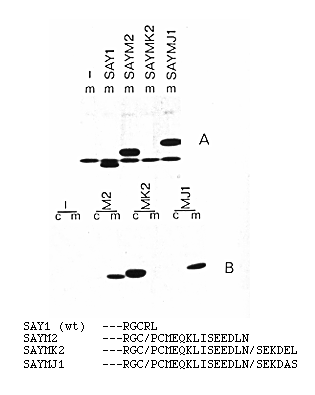
Figure 6. Expression of Lysozyme Derivatives
The carboxy-terminal amino acid sequences of the proteins encoded
by the various plasmids are shown at the bottom. (A) Medium from
[35S]Met labeled transfected cells. (B) Blot analysis
of cells and medium from cultures transfected with no DNA (-),
SAYM2 (M2), SAYMK2 (MK2), and SAYMJ1 (MJ1), probed with the anti-myc
monoclonal antibody Equivalent amounts of cells and medium were
analyzed.
The carboxy-terminal sequences of the proteins encoded by the various constructs are shown in Figure 6, together with the results of the transfection experiments. Figure 6A shows the analysis of [35S]Metlabeled proteins in the culture supernatants from transfected cells. Wildtype lysozyme, encoded by SAY1, was readily detectable in the medium; it migrates slightly faster on the gel than a prominent protein that is secreted by untransfected COS cells. SAYM2, which contains the c-myc tagging sequences, produced a correspondingly larger protein that was also efficiently secreted. Blotting analysis with the myc monoclonal antibody showed that almost all the SAYM2 protein made during a 3 hr incubation was present in the culture supernatant (Figure 6B). In contrast, the product of SAYMK2, which has both the myc sequence and the last six amino acids of grp78, was only just detectable in the supernatant (Figure 6A), and blotting showed that it had accumulated to very high levels in the cells (Figure 6B).
We were concerned that the addition of the grp78 sequences to lysozyme might interfere with its folding, and that this alone might be sufficient to prevent secretion. To minimize this possibility, a construct was made that was identical to SAYMK2, but had the last two codons of the grp78 sequence changed from Glu-Leu to Ala-Ser. This plasmid produced a protein that was secreted as efficiently as normal lysozyme (Figure 6: SAYMJ1). Replacement of the last six amino acids with five other unrelated heptapeptide sequences also failed to block secretion (data not shown; the peptides were SAEAARL, SREKAWL, SPNHSMV, SGPREWL, and SGPWEWL). These results argue strongly that the failure of the SAYMK2 protein to be secreted is not simply due to some problem of folding or aggregation, but is a specific consequence of the presence of the carboxy-terminal retention signal. The successful transfer of this signal to the lysozyme molecule indicates that no features of grp78 other than its carboxyl terminal sequence are required to prevent secretion.
The Retained Protein Is in the ER of Transfected Cells
If the signal identified in the previous experiments is solely
responsible for the intracellular location of luminal ER proteins,
then the lysozyme molecules bearing the grp78 carboxyl terminus
should accumulate in the ER and not in the Golgi or elsewhere
in the cell. To see whether this was true, we examined the intracellular
distribution of the various altered proteins by indirect immunofluorescence,
using the anti-myc monoclonal antibody. Untransfected cells
were not detectably stained with this antibody.
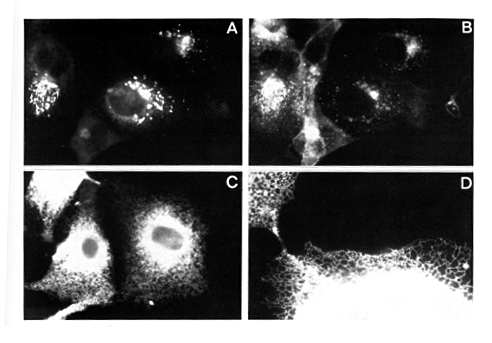
Figure 7 Immunofluorescent Staining of Transfected
COS Cells
(A and B) Cells transfected with SAYM2 (lysozyme tagged with the
c-myc peptide) double stained with the anti-myc
monoclonal (A) and rhodamine-conjugated wheat germ lectin (B).
The protein is mostly concentrated in the Golgi region, which
stains brightly with the wheat germ lectin. (C and D) Cells transfected
with SAYMK2 (same as SAYM2, but with six amino acids containing
the KDEL signal added) and stained with the anti-myc monoclonal.
(D) An enlargement of the periphery of a cell in which the tubular
ER network is clearly visible.
The lysozyme derivative containing the retention signal (SAYMK2) showed a completely different distribution (Figure 7C). At the periphery of the cells a network of tubules is clearly visible (Figure 7D). This pattern is very similar to that obtained when the ER is visualized with lipophilic fluorescent dyes (Pagano et al., 1981; Terasaki et al., 1984), which strongly suggests that the protein is indeed present in the ER. However, because of the bright staining in the perinuclear region, it was difficult to tell whether it is also present in the Golgi complex.
We were able to overcome the staining problem by using a confocal scanning laser microscope developed at the MRC Laboratory of Molecular Biology by John White (to be described elsewhere). This microscope uses a scanning laser beam to illuminate the specimen, thus reducing the glare from out-of-focus material and allowing the visualization of an optical section of the stained cells that is about 1 µm thick. Examples of the images obtained are shown in Figure 8. Figures 8A and 8B show the distribution of lysozyme without and with the retention signal, respectively. The images confirm the results obtained by conventional microscopy and allow detailed examination of the perinuclear distribution of the protein containing the retention signal (Figure 8B). There is no sign that this protein is concentrated in the Golgi.
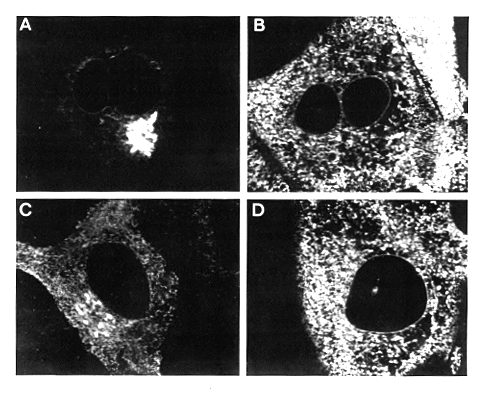
Figure 8. Stained COS Cells Viewed with the
Confocal Scanning Laser Microscope
All samples were stained with the anti-myc monoclonal,
and the microscope was focused about midway through the nucleus.
Cells were transfected with the following constructs: (A) SAYM2
(secretable lysozyme, equivalent to Figure 7A); (B) SAYMK2 (lysozyme
with the retention signal, equivalent to Figure 7C); (C) SAGM6
(grp78 with the retention signal deleted); (D) SAGMK1 (a derivative
of grp78 that retains the KDEL signal). Golgi staining is visible
in (A) and (C), but not in (B) or (D).
Figure 8C shows a cell transfected with SAGM6, which encodes a grp78 mutant lacking the carboxyterminal 15 amino acids. This protein is secreted much more slowly than lysozyme, and the ER staining is much more prominent than in Figure 8A, indicating that transport from the ER to the Golgi is rate-limiting. Nevertheless, Golgi staining is also clearly visible. In contrast, cells transfected with SAGMK1, a grp78 derivative containing the retention signal (Figure 2), show intense staining of the ER and nuclear envelope, but not of the Golgi (Figure 8D). An identical though fainter pattern was obtained when the monoclonal antibody described by Bole et al. (1986) was used to visualize the endogenous grp78 in COS cells (data not shown). Taken together, the immunofluorescent images strongly support the conclusion that the retention signal contained in the last six amino acids of grp78 prevents net movement of proteins from the ER to the Golgi.
Discussion
A Signal That Prevents Soluble ER Proteins from Being Secreted
We have shown that a short sequence at the carboxyl terminus of
GRP78 is both necessary to prevent loss of this protein from the
ER and sufficient to cause retention of a normally secreted protein
in the ER. Since there are presumably fewer types of soluble proteins
resident in the ER than there are secreted proteins, the existence
of a constitutive secretion mechanism and a specific retention
signal seems an economical way for cells to solve this particular
sorting problem. The signal itself has to be at the carboxyl terminus
and must comprise more than the terminal leucine residue but not
more than the last six residues of grp78. Comparison of the sequences
of different ER proteins suggests that the tetrapeptide Lys-AspGlu-Leu
(KDEL in the one-letter code) is the signal (Figure 1).
It is hard to rule out the possibility that changes in one region of a protein have a global effect on its folding and thus influence its behavior in an indirect fashion. Such problems can be minimized by comparing several different mutants, and by examining the effects of minimal changes. Five different alterations involving the KDEL sequence of grp78 caused its secretion, whereas a similar change that leaves KDEL intact did not. Only one of eight different additions to the carboxyl terminus of lysozyme resulted in its retention - the one that adds KDEL to the protein. In each case, changes involving only two amino acids were sufficient to convert a secreted protein to a retained one or vice versa. The simplest interpretation of these results is that the carboxyl terminus is recognized directly, and any indirect effects on protein folding are negligible. Although recognition of KDEL may well be influenced by its accessibility or by local features such as a net negative charge in the region, but it appears that the signal is a linear sequence rather than a complex three dimensional structure.
The absence of a KDEL signal does not guarantee that a protein will leave the ER, and those that do leave do so at very different rates (compare truncated grp78 and lysozyme in Figure 4 and Figure 6; for review see Gebhart and Ruddon, 1986). One reason why some are not transported efficiently may be that they bind to other proteins that have a retention signal. For example, a number of mutant or unfolded proteins are known to bind tightly to grp78 ("Binding Protein") (e.g., Gething et al., 1986; Sharma et al., 1985), and as we have suggested previously, this may provide a mechanism by which they are held in the ER until their assembly is complete (Pelham, 1986).
Our experiments do not address the question of how transmembrane proteins that are restricted to the ER are distinguished from membrane proteins destined for other cellular compartments. At least some of these proteins have their carboxyl termini on the cytoplasmic side of the ER membrane and thus cannot be sorted by the KDEL system (unless they bind to a protein in the lumen). Indeed, deletion of the membrane anchor results in secretion of two viral proteins that are normally found in the ER membrane, one of which has its carboxyl terminus in the lumen (Paabo et al., 1986; Poruchynsky et al., 1985). The existence of a cytoplasmic domain on such proteins and their restriction to a two-dimensional compartment may allow quite different sorting mechanisms to operate.
Possible Mechanisms tor Retention in the ER
What recognizes the KDEL sequence? One simple possibility would
be that the ER proteins that appear to be soluble when cells are
lysed are normally anchored to the membrane via an integral membrane
protein that binds the KDEL sequence. A precedent for this would
be the retention of ß-glucuronidase, a portion of which
is held in the ER by tight stoichiometric interaction with a membrane
protein termed egasyn (for references see Medda and Swank, 1985).
Such a mechanism seems unlikely for two reasons. First, although
the ß-glucuronidase/egasyn complex can readily be identified
by immunoprecipitation, no such complexes have been identified
for grp78, grp94, or protein disulphide isomerase; on the contrary,
they are readily soluble and rapidly released upon homogenization
of the ER (Bole et al., 1986; Koch et al., 1986; Freedman, 1984).
In the case of grp78, complexes with newly synthesized secreted
proteins (such as immunoglobulin heavy chains) can be found, but
no anchoring protein has been detected. Indeed, the postulated
roles of proteins such as grp78 and the disulphide isomerase in
protein assembly (Pelham, 1986; Freedman, 1984) may require that
they are free to reach their substrates in the lumen. Second,
grp78 and grp94 are two of the most abundant proteins in the ER,
and there is no obvious candidate for a protein that is sufficiently
abundant to bind all of them. Moreover, overproduction of grp78
by at least 7-fold does not result in its secretion from COS cells
(Figure 3). This apparent inability to saturate the retention
system argues against simple stoichiometric binding.
An alternative possibility might be that the KDEL sequence is recognized by an enzyme that modifies the protein in some way, for example by addition of a lipid molecule. Such catalytic action would be relatively resistant to saturation. However, the modified protein would still have to be sorted. Moreover, there is no evidence for such modification; the proteins do not appear membrane-bound, and the lysozyme derivative with KDEL at its carboxyl terminus, which is retained, has exactly the same mobility on SDS-polyacrylamide gels as the version with KDAS, which is secreted (Figure 6 and unpublished observations).
The data could be explained if the proteins were not held in the ER, but instead were continually retrieved from some later point on the secretion pathway and returned to it. The contents of the ER is thought to be delivered to the cis Golgi by a rapid and efficient process of vesicle budding and fusion (Palade, 1975). Since there is no evidence for membrane traffic to the ER from the trans side of the Golgi complex or from the plasma membrane, it seems likely that there is a reverse flow from the cis Golgi (or from something close to it in the pathway) to the ER. In principle, a specific receptor could recognize soluble ER proteins that had been delivered to the cis Golgi along with secreted proteins and carry them back to the ER. Such a receptor would bind KDEL in the Golgi environment, but not in the ER. It would be difficult to saturate because it would act catalytically and be concentrated in a relatively small area; if retrieval were efficient, there would be no accumulation of the proteins in the Golgi. This model is very similar to the one proposed by Rothman (1981) for sorting of ER membrane proteins.
Whatever the retention mechanism, there must be some cellular component that recognizes the KDEL sequence. It should be possible to test various sorting models by identifying and characterizing this component.
Experimental Procedures
Plasmids
The COS cell expression vectors are based on plasmid AH5. This
plasmid contains the SV40 origin, the adenovirus major late promoter,
and the Drosophila hsp70 gene between unique HindIII and Sall
sites. A derivative, AHP2, has the Sall site converted to an EcoRI
site (Munro and Pelham, 1984). SAG1 was tormed by insertion of
a Nael-Sspl fragment containing the complete grp78 coding sequence
from clone R76 (Munro and Pelham, 1986) between the HindIII and
EcoRI sites of AHP2, using intermediate cloning between the Pstl
and Xbal sites (blunted) of pUC12 to generate the appropriate
sites at either end of the gene (the XbaI site, at the 3' end
of the gene, was reformed). The gene was truncated in SAG4 by
fusion of the DraII site at amino acid 622 to the SalI site of
AH5.
The amino-terminal deletion in SIG2 was formed by replacing the fragment in SAG1 between the HindIII site and the FokI site at amino acid 13 with an AvaII-PstI fragment that encodes the leader sequence and first five amino acids of an immunoglobulin heavy chain (Neuberger, 1983). The PstlFokl junction contains sequences from the pUC12 polylinker (PstI to filled-in AccI). The aminoterminal region of SIGM2 is similar, but the polylinker-derived sequences are slightly different (blunted PstI to filled-in SalI), generating a different amino acid sequence at this point (see Figure 2).
SAGGL was made using a Bal31 deletion mutant of the grp78 cDNA generated during sequencing. This removed the termination codon, but a new one is formed by an adjacent polylinker-derived Xbal site. The mutated carboxyl terminus (KpnI-XbaI) was used to replace the corresponding region in SAG1.
Sequences encoding the c-myc epitope were originally defined by Bal31 deletion of the human c-myc gene and are flanked by a 5' NcoI site and a 3' EcoRI. Clones containing this sequence were constructed using a derivative of AHP2 from which other Ncol sites had been removed. In SAGM2, SIGM2, and SAGMK1 the NcoI site was joined to the NcoI site in the grp78 gene. In SAGM6 the (filled-in) Ncol site was first fused to a (filled in) SalI site, which was then cut, filled in, and fused to the (filled-in) DraII site at amino acid 622. In SAGM2, SIGM2, and SAGM6 the EcoRI site at the 3' end of the myc sequence was filled in to generate an in-frame termination codon. For SAGMK1, a doublestranded synthetic oligonucleotide with EcoRI sticky ends that encoded the last six amino acids of grp78 and a termination codon (AATTCGGAGAAGGATGAGCTCTAG/AATT) was cloned into the EcoRI site at the 3' end of the myc sequence.
The chick lysozyme cDNA sequence with a HindIII site introduced just upstream of the ATG was obtained from Alan Colman (Krieg et al., 1984). A HindIII-NaeI fragment encoding all but the last two amino acids of the protein was cloned between the HindIII and HincII sites of pUC19; this results in a sequence that fortuitously encodes the wildtype carboxyl terminus, termination occurring within the adjacent Xbal site. The HindIII-XbaI fragment was then used to replace the grp78 gene in SAG1, forming SAY1. SAYM2 was formed by inserting the original HindIII-NaeI fragment between the HindIII and NcoI (filled) sites of SAGM2, and then filling in the (reformed) NcoI site to adjust the reading frame. SAYMK2 was formed similarly from SAGMK1. The synthetic oligonucleotide (see above) encoding the SEKDEL sequence of SAYMK2 was designed such that the last two codons and the termination codon consist of overlapping SacI and Xbal sites. The carboxyl terminus of SAYMK1 was modified to form SAYMJ1 by filling in the XbaI site and then trimming the overhanging ends of the Sacl site with Klenow, which has the effect of changing the last two codons from GluLeu to Ala-Ser.
The amino acids encoded by the critical regions of these plasmids are shown in Figure 2 and Figure 6. Junction sequences and the orientation of synthetic oligonucleotide inserts were confirmed by restriction enzyme digestion and/or by DNA sequencing of appropriate plasmids.
Transfection of Cells and Analysis of Proteins
COS cells were transfected in the presence of 0.5 mg/ml DEAE-dextran,
followed by a 3 hr treatment with 100 µg/ml chloroquine
in complete medium, as previously described (Pelham, 1984). After
40-50 hr they were washed in serum-free medium and then incubated
in the absence of serum for 2-4 hr. For labeling, this incubation
was in methionine-free medium to which 100 µCi/ml [35S]Met
had been added. The medium was then removed from the cells, ovalbumin
was added to 30 µg/ml, and the protein was precipitated
with 10% trichloroacetic acid.
Samples of cells or culture medium were analyzed on SDS polyacrylamide gels and either fluorographed using Amplify (Amersham) or transferred to nitrocellulose for reaction with antibodies as previously described (Munro and Pelham, 1984). Usually equivalent amounts of the cells and culture medium were analyzed, but in Figures 3A and 3C, where labeling was for only 2 hr, five times as much material from the medium as from the equivalent cell pellet was loaded on the gel.
For immunoprecipitation, samples of culture medium or the postnuclear supernatant of Nonidet P-40 lysed cells were incubated with rabbit anti-grp78 serum. Immune complexes were recovered using protein A-Sepharose as described previously (Munro and Pelham, 1986).
Antibodies
Polyclonal rabbit anti-rat grp78 prepared against purified, denatured
protein was provided by M. Lewis. A mouse monoclonal antibody
(9E10) raised against a synthetic peptide comprising residues
409-439 of human c-myc (Evan et al., 1985) was a gift from
G. Evan. A rat monoclonal anti hsp70 (710), which cross-reacts
with denatured grp78 (see Munro and Pelham, 1986), was provided
by S. Lindquist. A mouse monoclonal that recognizes native (but
not denatured) chick egg-white lysozyme (D1.3; Mariuzza et al.,
1983) was a gift from J. Foote. A rat monoclonal antibody raised
against mouse grp78 (BiP) (Bole et al., 1986) was provided by
D. Bole. The anti-myc antibody was detected on blots with
125I-labeled F(ab')2 sheep anti-mouse 19 (Amersham).
For immunofluorescence, it was visualized with FlTC-labeled rabbit
antimouse IgG (Miles) that was affinity-purified by J. Kilmartin.
Wheat germ lectin (Pharmacia) was conjugated to rhodamine by M.
Robinson (Robinson and Pearse, 1986).
Immunofluorescence
Cells were transferred to slides and processed for immunofluorescence
as described previously (Pelham, 1984), except that the cells
were fixed with 2% paraformaldehyde/0.1% glutaraldehyde, permeabilized
in 1% Triton X-100 in phosphate-buffered saline, and treated for
10 min with 1 mg/ml NaBH4 (BDH, freshly dissolved in phosphate-buffered
saline). Inclusion of glutaraldehyde in the fixation proved essential
for good preservation of ER structure. Stained cells were photographed
on a Zeiss Standard microscope fitted with epifluorescent illumination,
using Kodak Technical Pan film. Images from the same slides were
generated by the confocal scanning laser microscope on a video
monitor, and this was photographed using Ilford FP4 film. The
laser microscope was designed by John White and built at the MRC
Laboratory of Molecular Biology; it will be described in detail
elsewhere (J. White and B. Amos, personal communication).
Acknowledgments
We thank Mike Lewis for much valuable assistance and for preparing
the anti-grp78 antiserum. We also thank all those who provided
monoclonal antibodies, Tony Hyman and John White for assistance
with the confocal laser microscope, Terry Smith for synthetic
oligonucleotides, and our colleagues at the MRC for much advice
and helpful discussion. We are grateful to lan Charles and Tom
Creighton for the plasmids encoding lysozyme with five different
heptopeptides at the carboxyl terminus.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received December 22, 1986.
References
Bole, D. G., Hendershot, L. M., and Kearney, J. F. (1986). Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J. Cell Biol. 102,15581566.
Cutler, D., Lane, C., and Colman, A. (1981). Non-parallel kinetics and the role of tissue-specific factors in the secretion of chicken ovalbumin and lysozyme from Xenopus oocytes. J. Mol. Biol. 153, 917-931.
Edman, J. C., Ellis, L., Blacher, R. W., Roth, R. A., and Rutter, W A. (1985). Sequence of protein disulphide isomerase and implications of its relationship to thioredoxin. Nature 317, 267-270.
Evan, G. I., Lewis, G. K., Ramsay, G., and Bishop, J. M. (1985). Isolation of monoclonal-antibodies specific for human c-myc proto- oncogene product. Mol. Cell. Biol. 5, 3610-3616.
Freedman, R. B. (1984). Native disulphide bond formation in protein biosynthesis: evidence for the role of protein disulphide isomerase. Trends Biochem. Sci. 9, 438-441.
Fries, E., Gustafsson, L., and Peterson, R A. (1984). Four secretory pro" teins synthesised by hepatocytes are transported from endoplasmic reticulum to Golgi complex at different rates. EMBO J. 3, 147-152.
Gebhart, A. M., and Ruddon, R. W. (1986). What regulates secretion of non stored proteins by eukaryotic cells? BioEssays 4, 213-218.
Gething, M. -J., McCammon, K., and Sambrook, J. (1986). Expression of wild-type and mutant forms of influenza hemagglutinin: the role of folding in intracellular transport. Cell 46, 939-950.
Haas, I. G., and Wabl, M. (1983). Immunoglobulin heavy chain binding protein. Nature 306, 387-389.
Kivirikko, K. I., and Myllyla, R. (1982). Posttranslational enzymes in the biosynthesis of collagen: intracellular enzymes. Meth. Enzymol. 82, 245 304.
Koch, G., Smith, M., Macer, D., Webster, P, and Mortara, R. (1986). Endoplasmic reticulum contains a common, abundant calcium-binding glycoprotein, endoplasmin. J. Cell Sci. 86, 217-232.
Kondor-Koch, C., Bravo, R., Fuller, S. D., Cutler, D., and Garoff, H. (1985). Exocytotic pathways exist to both the apical and the basolateral cell surface of the polarized epithelial cell MDCK. Cell 43, 297-306.
Krieg, P, Strachan, R., Wallis, E., Tabe, L., and Colman, A. (1984). Efficient expression of cloned complementary DNAs for secretory proteins after injection into Xenopus oocytes. J. Mol. Biol. 180, 615-643.
Kulomaa, M. S., Weigel, N. L., Kleinsek, D. A., Beattie, W. G., Conneely, O. M., March, C., ZaruckiSchulz, T., Schrader, W. T, and O'Malley, B. W (1986). Amino acid sequence of a chicken heat shock protein derived from the complementary DNA nucleotide sequence. Biochemistry 25, 6244-6251.
Lee, A. S., Bell, J., and Ting, J. (1984). Biochemical characterization of the 94- and 78-kilodalton glucose-regulated proteins in hamster fibroblasts. J. Biol. Chem. 259, 4616-4621.
Lewis, M. J., Mazzarella, R. A., and Green, M. (1985). Structure and assembly of the endoplasmic reticulum. The synthesis of three major endoplasmic reticulum proteins during lipopolysaccharideinduced differentiation of murine Iymphocytes. J. Biol. Chem. 260, 3050-3057
Mariuzza, R. A., Jankovic, D. LJ., Boulot, G., Amit, A. G., Saludjian, R, Le Guern, A., Mazie, J. C., and Poljak, R. J. (1983). Preliminary crystallographic study of the complex between the Fab fragment of a monoclonal anti-lysozyme antibody and its antigen. J. Mol. Biol. 170, 1055-1058.
Medda, S., and Swank, R. T (1985). Egasyn, a protein which deter" mines the subcellular distribution of beta-glucuronidase, has esterase activity. J. Biol. Chem. 260, 15802-15808.
Munro, S., and Pelham, H. R. B. (1984). Use of peptide tagging to detect proteins expressed from cloned genes: deletion mapping functional domains of Drosophila hsp70. EMBO J. 3, 3087-3093.
Munro, S., and Pelham, H. R. B. (1986). An hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 46, 291-300.
Neuberger, M. S. (1983). Expression and regulation of immunoglobulin heavy chain gene transfected into Iymphoid cells. EMBO J. 2, 1373-1378.
Paabo, S., Weber, F., Nilsson, T, Schaffner, W., and Peterson, R A. (1986). Structural and functional dissection of an MHC class I antigen" binding adenovirus glycoprotein. EMBO J. 5, 1921-1927
Pagano, R. E., Longmuir, K. J., Martin, O. C., and Struck, D. K. (1981). Metabolism and intracellular localization of a fluorescently labelled intermediate in lipid biosynthesis within cultured fibroblasts. J. Cell Biol. 91, 872-877
Palade, G. (1975). Intracellular aspects of the process of protein synthesis. Science 189, 347-358.
Pelham, H. R. B. (1984). Hsp70 accelerates the recovery of nucleolar morphology after heat shock. EMBO J. 3, 3095-3100.
Pelham, H. R. B. (1986). Speculations on the functions of the major heat shock and glucose-regulated proteins. Cell 46, 959-961.
Poruchynsky, M. S., Tyndall, C., Both, G. W., Sato, F, Bellamy, A. R., and Atkinson, R H. (1985). Deletions into a NH2-terminal hydrophobic domain result in secretion of Rotavirus VP7, a resident endoplasmic reticulum membrane glycoprotein. J. Cell Biol. 101, 2199-2209.
Robinson, M. S., and Pearse, B. M. F. (1986). Immunofluorescent localisation of 100K coated vesicle proteins. J. Cell Biol. 102, 48-54.
Rothman, J. E. (1981). The Golgi apparatus: two organelles in tandem. Science 213, 1212-1219.
Sharma, S., Rodgers, L., Brandsma, J., Gething, M. -J., and Sambrook, J. (1985). SV40 T antigen and the exocytic pathway. EMBO J. 4, 1479-1489.
Shiu, R. P C., Pouyssegur, J., and Pastan, I. (1977). Glucose
depletion
accounts for the induction of two transformation-sensitive membrane
proteins in Rous sarcoma virustransformed chick embryo fibroblasts.
Proc. Natl. Acad. Sci. USA 74, 3840-3844.
Sorger, R K., and Pelham, H. R. B. (1987). The glucose regulated protein grp94 is related to heat shock protein hsp90. J. Mol. Biol., in press.
Terasaki, M., Song, J., Wong, J. R., Weiss, M. J., and Chen, L. B. (1984), Localization of endoplasmic reticulum in living and glutaraldehyde-fixed cells with fluorescent dyes. Cell 38, 101-108.
Virtanen, I., Ekblom, R, and Laurila, R (1980). Subcellular compartmentalization of saccharide moieties in cultured normal and malignant cells. J. Cell Biol. 85, 429-434.
Welch, W J., Garrels, J. I., Thomas, R G., Lin, J. J. C., and Feramisco, J. R. (1983). Biochemical characterization of the mammalian stress proteins and identification of two stress proteins as glucoseand Ca2+-ionophore regulated proteins. J. Biol. Chem. 258, 7102-7111.
![]()
![]()
©
Copyright 2000 Department of Biology, Davidson College, Davidson,
NC 28036
Send comments, questions, and suggestions to: macampbell@davidson.edu